doi: 10.62486/agmu202463
ORIGINAL
Clinical diagnosis of chronic mucocutaneous candidiasis in a patient with recurrent staphylococcal infections
Diagnóstico clínico de candidiasis mucocutánea crónica en un paciente con infecciones estafilocóccicas recurrentes
Luis Alexis Peláez Yáñez1 *, Odalys Orraca Castillo1, Jessica O. Solana Rodríguez1, Mayelín García García1, Julio Israel Hernández Pacheco1
1Universidad de Ciencias Médicas de Pinar del Río. Hospital Pediátrico Provincial “Pepe Portilla”, Servicio de Inmunología. Pinar del Río. Cuba.
Citar como: Peláez Yáñez LA, Orraca Castillo O, Solana Rodríguez JO, García García M, Hernández Pacheco JI. Clinical diagnosis of chronic mucocutaneous candidiasis in a patient with recurrent staphylococcal infections. Multidisciplinar (Montevideo). 2024; 2:63. https://doi.org/10.62486/agmu202463
Enviado: 20-11-2023 Revisado: 17-04-2024 Aceptado: 11-09-2024 Publicado: 12-09-2024
Editor: Telmo
Raúl Aveiro-Róbalo ![]()
Autor para la correspondencia: Luis Alexis Peláez Yáñez *
ABSTRACT
Chronic mucocutaneous candidiasis is a primary immunodeficiency characterized by persistent or recurrent candidal infections of the skin, nails, or mucous membranes. It can be associated with endocrinopathies and autoimmune diseases. A 37-years-old male patient is presented who attended an immunology consultation referred from the dermatology service due to recurrent pyodermitis of several years of evolution; the physical examination also revealed signs of candidiasis in the oral mucosa and lip corners, in the eyelids of both eyes and skin in the abdominal region, which were recurrent, as well as onychomycosis in all toenails. The microbiological study of the pyodermitis lesions showed infection by staphylococcus aureus, and the lesions of the mouth, eyes, abdominal skin and nails showed the presence of Candida albicans. Delayed intradermal skin testing was performed with no T cell response to Candida and Trichophyton antigens. Complementary drugs were indicated to rule out any associated endocrine disorder.
Keywords: Chronic Mucocutaneous Candidiasis; Primary Immunodeficiency; Recurrent Pyodermitis.
RESUMEN
La candidiasis mucocutánea crónica (CMC) es una inmunodeficiencia primaria que se caracteriza por infecciones candidiásicas persistentes o recurrentes en piel, uñas o membranas mucosas. Puede asociarse con endocrinopatías y con enfermedades autoinmunitarias. Se presenta un paciente masculino de 37 años de edad que asistió a consulta de inmunología remitido desde el servicio de dermatología por presentar piodermitis recurrentes de varios años de evolución; al examen físico se le constató, además, signos de candidiasis en mucosa oral y comisuras labiales, en párpados de ambos ojos y piel de región abdominal, recidivantes, así como onicomicosis en todas las uñas de los pies. El estudio microbiológico de las lesiones por piodermitis arrojó infección por estafilococo dorado, y el de las lesiones de la boca, ojo y piel abdominal y uñas mostraron la presencia de Candida albicans. Se realizaron pruebas cutáneas demoradas intradérmicas sin respuesta de células T a los antígenos de Cándidas y Trichophyton. Se indicaron complementarios para descartar algún trastorno endocrino asociado.
Palabras clave: Candidiasis Mucocutánea Crónica; Inmunodeficiencia Primaria; Piodermitis Recidivante.
INTRODUCCIÓN
La CMC comprende un grupo de enfermedades con gran heterogeneidad clínica que se caracterizan por infecciones recurrentes por Candida albicans en piel, uñas y mucosas;(1,2,3) se clasifica dentro de los errores innatos de la inmunidad.(4,5)
La enfermedad aparece frecuentemente en la infancia aunque puede ser en la edad adulta. Los pacientes en raras ocasiones desarrollan infecciones sistémicas por Cándida, pero tienen susceptibilidad mayor que individuos inmunocompetentes para infecciones por bacterias encapsuladas y presencia de infecciones respiratorias. Las complicaciones severas se desarrollan en raras ocasiones pero se han descrito infecciones por Histoplasma capsulatum, Criptococcus neoformans, o Nocardia. Puede asociarse con endocrinopatías y con enfermedades autoinmunitarias.(2,4,5)
La mayoría de los casos son esporádicos, pero existen numerosos reportes familiares. Se han observado patrones de herencia autosómicos dominantes y recesivos.(4)
La inmunidad mediada por células es esencial en la respuesta inmunitaria contra Cándida sp; investigaciones recientes detectaron diversos defectos inmunológicos en IL-17 y sus receptores, mutaciones en el factor de transductor de señal y activador de la transcripción (STAT 1) y defectos ya demostrados en el eje IL-12-interferón gamma.(6,12)
PRESENTACIÓN DE UN CASO
Paciente masculino, de 37 años de edad, de raza blanca con antecedentes patológicos personales de procesos infecciosos a repetición; con historia de infecciones estafilocóccicas frecuentes dadas por forunculosis y abscesos recidivantes con necesidad de antibioticoterapia, inefectiva por las recurrencias de éstas, por las cuales se interconsulta con nuestro servicio de inmunología.
Al realizarle historia clínica encontramos que desde muy pequeño padecía de infecciones respiratorias como cuadros catarrales y otitis medias supurativas y también de infecciones candidiásicas de mucosa oral y de piel. Ya después de un año de edad comenzó a presentar infecciones de piel de etiología estafilocóccica; en varias ocasiones presentó cuadros diarreicos desde su niñez. Tuvo cuadros de sinovitis de rodilla en muchas ocasiones. Hasta ese momento de la primera consulta con nuestro servicio el paciente mantenía lesiones en ambas comisuras labiales y en piel de región abdominal, y onicomicosis en todas las uñas de ambos pies. Es de señalar, también, la presencia de lesiones por verrugas vulgares en ambas manos.
Este paciente necesitó de tratamientos en salas de cuidados intensivos; a sus 19 años de edad ingresó en servicio de cuidados intensivos del Instituto de Medicina Tropical “Pedro Kourí” por presentar criptococosis para lo cual llevó tratamiento y donde, además fue necesario realizarle esplenectomía “por presentar cifras muy bajas de hemoglobina y de plaquetas” (referido por el paciente); a los 27 años de edad, en la unidad de cuidados intensivos del Hospital Clínico Quirúrgico “Abel Santamaría” de Pinar del Río a causa de neumonía bacteriana con infección nosocomial sobreañadida de partes blandas por Pseudomona aeruginosa donde recibió tratamiento con varios antibióticos tales como vancomicina, meropenem y piperacilina con tazobactam.
Acudió a nuestra consulta con lesiones de piel dadas por presencia de forúnculo en región anterior interna medial del muslo derecho y con lesiones amplias eritematosas ligeramente escamosas localizadas en región abdominal ventral media e inferior con continuidad a espalda y regiones inguinales y glúteas, más llamativas a nivel de sus pliegues, asociadas a prurito intenso. Las uñas de todos los dedos de los pies están engrosadas y distróficas. (figura 1)
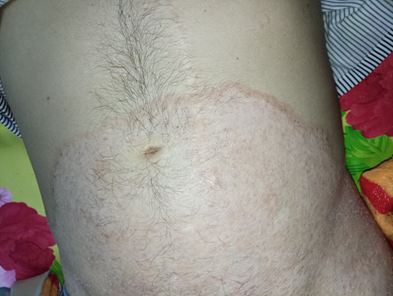
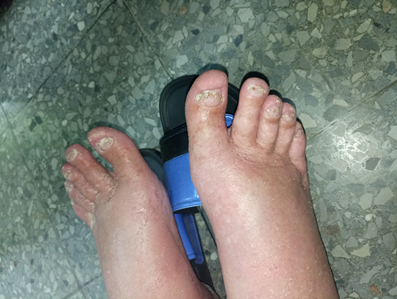
Figura 1. Lesiones amplias eritematosas ligeramente escamosas localizadas en región abdominal ventral media e inferior con continuidad a espalda y regiones inguinales y glúteas. Las uñas de todos los dedos de los pies están engrosadas y distróficas
Este paciente desde entonces se mantuvo bajo régimen de seguimiento estricto por nuestra consulta de inmunología donde se le realizaron diferentes estudios e indicó conducta terapéutica respectiva.
Se realizó exudado microbiológico de forúnculo con cultivo y antibiograma, así como raspado de piel y uñas afectadas con cultivo y antifungigrama; estos complementarios arrojaron la presencia de S. aureus con sensibilidad a cotrimoxazol, y la presencia de Candida albicans con sensibilidad positiva a todos los antimicóticos evaluados, respectivamente.
Se realizó prueba cutánea demorada, iintradermorreacción a candidina y tricofitina: negativas a las 24, 48 y 72 horas, lo que orientó el defecto en la respuesta celular mediada por linfocitos T a Candida sp, fundamentalmente.
Otros complementarios realizados: leucogramas con diferencial con valores normales de leucocitos con cantidades de las diferentes células blancas dentro de los límites de normalidad; lámina periférica, normal;los estudios de hormonas tiroideas T3 y T4 así como de TSH arrojaron valores normales; pruebas de detección de antígeno de superficie de la hepatitis B y anticuerpos anti C, negativas; test de VIH por ELISA, negativo; además de pruebas de proteína C reactiva, inmunocomplejos circulantes y factor reumatoideo, negativos; test VIH por ELISA, negativo y dosificación de inmunoglobulinas séricas y complemento con resultados normales:
IgG: 15,09g/L(6,80-14,45), IgA: 1,94 g/L(0,83-4,06), IgM: 1,26 g/L(0,34-2,14), IgE 62 UI/L (≤150), C3: 1,12 g/L(0,75-1,35), C4: 0,16 g/L(0,09-0,36).
El estudio hepático mostró valores normales de TGP y TGO con cifras menores de 25 unidades por mililitro; la ecografía señaló normalidad hepática ultrasonográfica.
Se inició tratamiento oral combinado de cotrimoxazol y rifampicina así como la aplicación de ungüento de neobatin en fosas nasales y a convivientes logrando respuesta clínica completa de la forunculosis a los diez días de iniciado el tratamiento antibacteriano.
Posteriormente al tratamiento antibacteriano se comenzó con tratamiento antimicótico local y sistémico, fluconazol (150mg) 1 cáp diaria por 10 días, con mejoría clínica, y que continuó posteriormente con esta misma dosis con frecuencia semanal durante seis meses en total, logrando mejoría casi total de las lesiones de piel y uñas.
Es de destacar que desde el inicio del tratamiento del paciente se indicó inmunoestimulación con factor de transferencia y sulfato de cinc así como vitaminoterapia.
DISCUSIÓN
Se presenta un paciente con CMC sin respuesta a la inoculación intradérmica de antígenos micóticos y con otras enfermedades asociadas. La presentación clínica de la CMC es muy variada dada la heterogeneidad de esta inmunodeficiencia primaria.(2,4) Lo que sugiere que bajo el denominador clínico antes descrito se agrupan diversas enfermedades específicas.(5)
Entre el 60 y el 80 % de los pacientes con CMC manifiesta su enfermedad en la infancia, sólo una minoría la expresa tardíamente.(3,7,13) Predomina la herencia autosómica recesiva y es menos frecuente la herencia autosómica dominante. En una minoría de los casos se presenta en forma esporádica; puede acompañarse de endocrinopatías como hipotiroidismo, hipoparatiroidismo, enfermedad de Addison, diabetes mellitus y de procesos autoinmunes como vitiligo, sarcoidosis, anemia perniciosa y anemia hemolítica autoinmune,(4,7,14) además de que se ha asociado a timoma en pacientes adultos.(7)
El síndrome de poliendocrinopatía autoinmunitaria-candidiasis-distrofia ectodérmica o APECED (por su sigla en inglés), también conocido como el síndrome poliglandular autoinmunitario de tipo 1,(15) se transmite con un patrón de herencia autosómica recesiva y es producto de mutaciones en el gen regulador autoinmunitario (AIRE).(7,16) Se caracteriza por la asociación de por lo menos dos de las siguientes enfermedades: CMC, hipotiroidismo y enfermedad de Addison.(16) Otras manifestaciones menos frecuentes de la APECED, son la tiroiditis autoinmunitaria, insuficiencia gonadal, anemia perniciosa, diabetes mellitus tipo 1, hepatitis autoinmunitaria, diarrea crónica, malabsorción y manifestaciones de distrofia ectodérmica: queratitis, alopecia, hipoplasia del esmalte dental y distrofia ungueal.(5) Manifestaciones clínicas no presentes en este paciente hasta este momento de su seguimiento.
Dentro de los 10 grupos de la clasificación de los errores innatos de la inmunidad,(17) este caso se corresponde con mayor posibilidad el grupo VI (defectos de la inmunidad innata e intrínseca), dado por las manifestaciones infecciosas a repetición con susceptibilidad a infecciones micóticas, estafilocóccicas y por pseudomonas que ha presentado. Se descartan las enfermedades del grupo IV (enfermedades inmunes por disregulaciones) que se caracterizan por infecciones micóticas pero el paciente no presenta manifestaciones de disregulación como son las poliendocrinopatías, típicas de estas entidades.(17)
Entre las enfermedades del grupo VI se descartan aquellas con herencia autosómica recesiva puesto que los padres del paciente no tienen iguales apellidos y proceden de zonas geográficas diferentes y distantes, por lo que se descarta la consanguinidad; con la posibilidad de un patrón de herencia autosómica dominante.(17) Al parecer se presenta en este caso una nueva mutación del gen, no se observan casos similares en la ascendencia de su familia ni en la familia de su esposa, y a su vez, éste tiene dos hijas, una de ellas sana y otra enferma, pudiéndose corroborar la etiología genética que se sospecha.
Dentro de las enfermedades autosómicas dominantes del grupo VI (defectos en la inmunidad innata e intrínseca), por las manifestaciones clínicas descritas, se sospecha una deficiencia de IL-17F,(6) que se presenta con un patrón autosómico dominante(10) y se caracteriza por foliculitis y candidiasis mucocutánea crónica,(8) lo cual se corresponde con el cuadro clínico del paciente.(17)
La IL-17 es una citocina proinflamatoria secretada principalmente por células T activadas. Presenta varias isoformas (A-F).(3,18) La IL-17ª es una citocina característica de las células Th17, aunque puede ser producida por otras líneas celulares como las células T ϒծ, NKT y TCD8+. Esta citocina favorece la diferenciación al patrón TH17 en la defensa frente a bacterias extracelulares y hongos, así como la migración de neutrófilos. Entre los miembros de la familia de la IL-17, la IL-17F presenta la homología más alta con la IL-17ª (60 %).(3) Los genes que codifican para los homodímeros IL-17ª e IL-17F de la citocina IL-17, se encuentran agrupados en el cromosoma 14A en el ratón y en el 6pR en el humano.(6) La IL-17ª y la IL-17F pueden ser secretadas como homodímeros y heterodímeros,(8) siendo el heterodímero IL-17A/F más potente que la IL-17A en la inducción de la expresión de quimiocinas. Esta citocina estimula varias células para la producción de mediadores inflamatorios IL-1, TNFα y otras citocinas, necesarios para el reclutamiento de neutrófilos y otros leucocitos, evento característico de la enfermedad inflamatoria.(6,8)
La pérdida de función de STAT1 es otra enfermedad autosómica dominante dentro de este grupo que pudiera presentar el paciente que se discute.(9,10,11,12) Se caracteriza por la susceptibilidad a infecciones por hongos, bacterias, virus y autoinmunidad, por enteropatías y manifestaciones endocrinas como diabetes mellitus y tiroiditis(8,19,20) que no están presentes en el paciente hasta este momento, por lo que queda descartada.
El manejo de los pacientes con candidiasis mucocutánea crónica se basa en el tratamiento de las enfermedades infecciosas secundarias al déficit inmunológico y de las endocrinopatías y complicaciones asociadas.(2) Una de las opciones coadyuvantes para el tratamiento de la candidiasis mucocutánea crónica es el factor de transferencia o extracto dializable de leucocitos,(2) ya que se reportó mejoría clínica de los pacientes con esta afección durante el tratamiento con factor de transferencia,(7) el mismo que se aplicó con buena respuesta en este caso y donde hasta ahora el paciente no ha presentado nuevas complicaciones infecciosas después de nuestro seguimiento y tratamiento.
CONCLUSIONES
Actualmente se tiene mayor reconocimiento de los trastornos monogénicos asociados con una susceptibilidad incrementada a infecciones fúngicas. Los descubrimientos hasta ahora realizados indican dónde se ubica el conocimiento acerca de los errores innatos de la inmunidad y orientan en la ruta del análisis de los trastornos inmunitarios por inmunodeficiencias primarias.
REFERENCIAS
1. García P, Aquino L, Vega DC, Ortega F, Arenas R. Candidiasis mucocutánea crónica en un paciente con síndrome poliendrocrino autoinmunitario tipo 1. Med Interna Mex. 2022;38(6):1274,1278.
2. Santa Elena AM, Pantoja A, Cisneros N, Addine B de la C, Parada ME. Diagnóstico y evolución de un caso de candidiasis mucocutánea crónica. Available from: http://www.cibamanz2020.sld.cu/index.php/cibamanz/cibamanz2020/paper/view/114/81
3. Venturi P, González A, Carolina AM, Marín C, Angiolini SC, Sotomayor CE. Estudio del perfil Th17 en pacientes pediátricos con candidiasis mucocutánea crónica de la provincia de Mendoza. Arch Alerg e Inmunol Clínica [Internet]. 2022;53:145,156. Available from: https://www.researchgate.net/profile/Claudia-Sotomayor-5/publication/378139417_Th17_profile_in_chronic_mucocutaneous_candidiasis_pediatric_patients_in_Mendoza_province/links/65c8e6cb1e1ec12eff81acac/Th17-profile-in-chronic-mucocutaneous-candidiasis-pediat
4. Ramírez BG, Sáez de Ocaris M del M. Aspectos diagnósticos, fisipatológicos y clínicos de la candidiasis mucocutánea crónica. Revisión de la literatura [Internet]. 2011. Available from: http://repositorio.pediatria.gob.mx:8180/hadle/20.500.12103/1175
5. Egri N, Esteve-Solé A. Deyà-Martínez À, Ortiz de Landazuri I, Vlagea A, García AP. Primary immunodeficiency and chronic mucocutaneous candidiasis: pathophysiological, diagnostic, and therapeutic approaches. Rev Allergol Immunopathol (Madr). 2021;49(1):118-27.
6. Chimenez R, Tropeano A, Chirico V, Ceravolo G, Salpietro C, Cuppari C. IL-17 serum level in patients with chronic mucocutaneous candidiasis disease. Rev Pediatr Allergy Immunol. 2022;33(27):77-9.
7. Chávez MC, Marcelo JL, Gómez Y, González A, Pérez T. Candidiasis mucocutánea crónica. A propósito de un caso. Revista cubana de hematología, inmunología y hemoterapia. 2017;36(suplemento):1-6.
8. Flores C, Bonifaz A. Aspectos inmunológicos de la candidosis mucocutánea crónica. Dermatología Cosmética, Médica y Quirúrgica [Internet]. 2020;18(4):296–305. Available from: https://www.medigraphic.com/pdfs/cosmetica/dcm-2020/dcm204l.pdf
9. Blanco Lobo P, Lei WT, Pelham SJ, Guisado Hernández P, Villaoslada I, de Felipe B. Biallelic TRAF3IP2 variants causing chronic mucocutaneous candidiasis in a child harboring a STAT1 variant. Rev Pediatr Allergy Immunol. 2021;32(8):1804-12.
10. Ostadi V, Sherkat R, Migaud M, Modaressadeghi SM, Casanova JL, Puel A. Functional analysis of two STAT1 gain-of-function mutations in two Iranian families with autosomal dominant chronic mucocutaneous candidiasis. Rev Med Mycol. 2021;59(2):180-8.
11. Carey B, Lambourne J, Porter S, Hodgson T. Chronic mucocutaneous candidiasis due to gain-of-function mutation in STAT1. Rev Oral Dis. 2019;25(3):684-92.
12. Baghad B, Benhsaien I, El Fatoiki FZ, Migaud M, Puel A, Chiheb S. [Chronic mucocutaneous candidiasis with STAT1 gain-of-function mutation associated with herpes virus and mycobacterial infections]. / Candidosecutanéo-muqueusechronique avec mutation gain-de-fonction du gène STAT1 associée à des infections herpétiqueset à mycobactérie. Ann Dermatol Venereol. 2020;147(1):41-5.
13. Frías MG, Rodríguez AC, Torres OU, Acosta G, Martínez E. Epidemiological data of candidiasis in a tertiary hospital in the State of Mexico. Dermatol Rev Mex. 2020;64(2):109-14.
14. García M, Pérez HF, Rojas CM, León X, Llausás E, García C. Chronic mucocutaneous candidiasis associated with autoimmunity and ectodermal dysplasia. A case report. Rev Alerg Mex. 2021;68(2):144-9.
15. Fierabracci A, Arena A, Toto F, Gallo N, Puel A, Migaud M. Autoimmune polyendocrine syndrome type 1 (APECED) in the Indian population: case report and review of a series of 45 patients. Rev J Endocrinol Invest. 2021;44(4):661-77.
16. Candelo PA, Arias DA, Victoria J. Candidiasis mucocutánea generalizada en una paciente con sindrome poliglandular tipo 1. A propósito de un caso. Rev Argentina Dermatología [Internet]. 2019;100. Available from: http://www.scielo.org.ar/scielo.php?script=sci_arttext&pid=S1851-300X2019000300101
17. Tangye SG, Al W, Aziz H, Charlotte B, Rundles C, Luis J. Human Inborn Errors of Immunity : 2022 Update on the Classification from the International Union of Immunological Societies Expert Committee Internet. Journal of Clinical Immunology. Springer US; 2022. 1473-1507 p. Available from: https://doi.org/10.1007/s10875-022-01289-3.
18. Verda S, Mirza D, Mallory L, MD Z, Steven R. Inmunidad al error innato de la vía inmunitaria Th17 asociada con la candidiasis mucocutánea crónica: una revisión sistemática. Rev Medicam en dermatología. 2023;22:1197–203.
19. Jobim M, Puel A, Ewald G, Gil BC, Migaud M, Fagundes IS, et al. Nova mutação no gene STAT1 associada com candidíase mucocutânea crônica. Arq Asmas Alerg e Imunol. 2020;4(3):354–9.
20. Chebli de Abreu, Nathalia Duarte S, Hyllo MJ, Neto A. Caso para el diagnóstico. Placas eritematosas y descamativas diseminadas: candidiasis mucocutánea crónica. Sci Direct [Internet]. 2023;98:691–4. Available from: https://www.sciencedirect.com/science/article/pii/S0365059623001101?via%3Dihub
FINANCIAMIENTO
Los autores no recibieron financiación para el desarrollo de la presente investigación.
CONFLICTO DE INTERÉS
Los autores declaran que no existe conflicto de intereses.
CONTRIBUCIONES DE LOS AUTORES
Conceptualización: Luis Alexis Peláez Yáñez, Odalys Orraca Castillo, Jessica O. Solana Rodríguez, Mayelín García García, Julio Israel Hernández Pacheco.
Curación de datos: Luis Alexis Peláez Yáñez, Odalys Orraca Castillo, Jessica O. Solana Rodríguez, Mayelín García García, Julio Israel Hernández Pacheco.
Análisis formal: Luis Alexis Peláez Yáñez, Odalys Orraca Castillo, Jessica O. Solana Rodríguez, Mayelín García García, Julio Israel Hernández Pacheco.
Investigación: Luis Alexis Peláez Yáñez, Odalys Orraca Castillo, Jessica O. Solana Rodríguez, Mayelín García García, Julio Israel Hernández Pacheco.
Metodología: Luis Alexis Peláez Yáñez, Odalys Orraca Castillo, Jessica O. Solana Rodríguez, Mayelín García García, Julio Israel Hernández Pacheco.
Administración del proyecto: Luis Alexis Peláez Yáñez, Odalys Orraca Castillo, Jessica O. Solana Rodríguez, Mayelín García García, Julio Israel Hernández Pacheco.
Recursos: Luis Alexis Peláez Yáñez, Odalys Orraca Castillo, Jessica O. Solana Rodríguez, Mayelín García García, Julio Israel Hernández Pacheco.
Software: Luis Alexis Peláez Yáñez, Odalys Orraca Castillo, Jessica O. Solana Rodríguez, Mayelín García García, Julio Israel Hernández Pacheco.
Supervisión: Luis Alexis Peláez Yáñez, Odalys Orraca Castillo, Jessica O. Solana Rodríguez, Mayelín García García, Julio Israel Hernández Pacheco.
Validación: Luis Alexis Peláez Yáñez, Odalys Orraca Castillo, Jessica O. Solana Rodríguez, Mayelín García García, Julio Israel Hernández Pacheco.
Visualización: Luis Alexis Peláez Yáñez, Odalys Orraca Castillo, Jessica O. Solana Rodríguez, Mayelín García García, Julio Israel Hernández Pacheco.
Redacción – borrador original: Luis Alexis Peláez Yáñez, Odalys Orraca Castillo, Jessica O. Solana Rodríguez, Mayelín García García, Julio Israel Hernández Pacheco.
Redacción – revisión y edición: Luis Alexis Peláez Yáñez, Odalys Orraca Castillo, Jessica O. Solana Rodríguez, Mayelín García García, Julio Israel Hernández Pacheco.