doi: 10.62486/agmu202467
ORIGINAL
Screening and diagnostic algorithm of hereditary metabolic nephropathies in newborns
Cribado y algoritmo diagnóstico de nefropatías metabólicas hereditarias en recién nacidos
Yangel
Fuentes Milián1 ![]() , Danyer Daniel Tamayo Ribeaux2
, Danyer Daniel Tamayo Ribeaux2
![]() ,
Anabel Cepero Rodríguez3
,
Anabel Cepero Rodríguez3 ![]() ,
Bárbara Martínez Pérez4
,
Bárbara Martínez Pérez4 ![]()
1Universidad de Ciencias Médicas de Pinar del Río. Hospital General Docente “Abel Santamaría Cuadrado”. Servicio de Nefrología. Pinar del Río, Cuba.
2Universidad de Ciencias Médicas de Granma. Facultad de Ciencias Médicas “Celia Sánchez Manduley”, Hospital Clínico-Quirúrgico Docente “Celia Sánchez Manduley”, Servicio de Medicina Interna. Granma, Cuba.
3Universidad de Ciencias Médicas de Ciego de Ávila. Facultad de Ciencias Médicas “José Assef Yara”, Departamento de Ciencias Básicas Biomédicas. Ciego de Ávila, Cuba.
4Universidad de Ciencias Médicas de Guantánamo. Facultad de Tecnología y Enfermería “Rafael García Moreaux”, Hospital General Docente “Dr. Agustino Neto”, Servicio de Laboratorio Clínico. Guantánamo, Cuba.
Citar como: Fuentes Milián Y, Tamayo Ribeaux DD, Cepero Rodríguez A, Martínez Pérez B. Screening and diagnostic algorithm of hereditary metabolic nephropathies in newborns. Multidisciplinar (Montevideo). 2024; 2:67. https://doi.org/10.62486/agmu202467
Enviado: 22-11-2023 Revisado: 16-03-2024 Aceptado: 30-08-2024 Publicado: 31-08-2024
Editor: Telmo Raúl
Aveiro-Róbalo ![]()
*Autor para la correspondencia: Yangel Fuentes Milián *
ABSTRACT
Introduction: inborn errors of metabolism expressed as hereditary nephropathies, entail various biochemical abnormalities that facilitate their screening and diagnosis in the newborn.
Objective: to offer a useful, ideal, simple and reliable screening alternative as a tool for the diagnosis of hereditary metabolic nephropathies in newborns.
Methods: an observational and cross-sectional study was carried out during the period September 2021-February 2023, at the Abel Santamaría Cuadrado General Teaching Hospital, Pinar del Río province, Cuba. The universe consisted of 90 patients and a representative sample of 63 was taken. The variables were studied: glycemia, lactinemia, ammonemia, arterial hemogasometry, urinalysis, hyperazotemia, heel test and perinatal risk factors associated with hyperazotemia. Empirical, theoretical and statistical methods were used. Medical ethics were respected.
Results: the correlation predominated hypoglycemia, hyperlactinemia and hyperammonemia with an incidence of 55,56 % and patients with metabolic acidosis in 49,21 %. A greater frequency was observed in the correlation of patients with alterations in the urinary supernatant and hyperazoemia, for 33,33 % of the sample. The number of patients with negative neonatal screening was higher, at 87 %. Low birth weight and prematurity were the perinatal risk factors most associated with hyperazoemia in the patients treated, 36,51 % and 33,33 % respectively.
Conclusions: the results obtained show high sensitivity and low specificity, but they still give us a reliable parameter and a tool to help diagnose hereditary metabolic nephropathies.
Keywords: Inborn Errors of Metabolism; Screening; Nephropathies; Newborn.
RESUMEN
Introducción: los errores innatos del metabolismo expresados como nefropatías hereditarias, conllevan
diversas anormalidades bioquímicas que facilitan su cribado y diagnóstico en el recién nacido.
Objetivo: ofrecer una alternativa de cribado útil, idónea, sencilla y confiable como herramienta para el diagnóstico de nefropatías metabólicas hereditarias en recién nacidos.
Métodos: se realizó un estudio observacional y transversal durante el periodo septiembre 2021-febrero 2023, en el Hospital General Docente Abel Santamaría Cuadrado, provincia Pinar del Río, Cuba. El universo estuvo constituido por 90 pacientes y se tomó una muestra representativa de 63. Se estudiaron las variables: glicemia, lactinemia, amonemia, hemogasometría arterial, uroanálisis, hiperazoemia, prueba del talón y factores de riesgo perinatales asociados a hiperazoemia. Se emplearon métodos empíricos, teóricos y estadísticos. Se respetó la ética médica.
Resultados: predominó la correlación hipoglicemia, hiperlactinemia e hiperamonemia con una incidencia del 55,56 % y los pacientes con acidosis metabólica en un 49,21 %. Se observó una mayor frecuencia en la correlación de pacientes con alteraciones en el sobrenadante urinario e hiperazoemia, para un 33,33 % de la muestra. Fue superior el número de pacientes con pesquisa neonatal negativa, para un 87 %. El bajo peso al nacer y la prematuridad fueron los factores de riesgo perinatales que más se asociaron a hiperazoemia en los pacientes atendidos para un 36,51 % y 33,33 % respectivamente.
Conclusiones: los resultados obtenidos evidencian una alta sensibilidad y una baja especificidad, pero aun así nos dan un parámetro confiable y una herramienta en la ayuda del diagnóstico de nefropatías metabólicas hereditarias.
Palabras clave: Errores Innatos del Metabolismo; Cribado; Nefropatías; Recién Nacido.
INTRODUCCIÓN
Los errores innatos del metabolismo intermedio (EIM) son un grupo heterogéneo de enfermedades monogénicas pertenecientes al grupo de las enfermedades raras (ER), que afectan vías metabólicas de la síntesis o catabolismo de los hidratos de carbono, aminoácidos o lípidos. Estas enfermedades conllevan anormalidades bioquímicas que pueden cuantificarse en diferentes fluidos biológicos, lo que permite su diagnóstico.(1,2,3)
Los Errores Innatos del Metabolismo (EIM), en su mayoría, tienen herencia autosómica recesiva (AR) y un grupo muy pequeño tienen una herencia recesiva ligada al cromosoma X. Esta situación, hace difícil advertir el nacimiento de pacientes con esta patología pues la mayor parte de veces, nacen de familias sanas, sin ningún antecedente de enfermedad metabólica.(4)
Los desórdenes metabólicos pueden ser causados por diversos mecanismos de deficiencia enzimática:
a) Interrupción de un paso de una vía metabólica produciendo acumulación de los metabolitos presentes antes de esta reacción enzimática. (Fenilcetonuria).
b) Inhabilidad de producir ciertos intermediarios o productos finales de una vía metabólica específica, como por ejemplo, cetoacidosis en la fase de ayuno en la deficiencia de la cadena media de la Acil-CoA deshidrogenasa (MCAD).
c) Alteraciones de más de una enzima que afectan varios pasos metabólicos (menos frecuentes), por ejemplo: deficiencia de múltiples sulfatasas o cofactores.(5)
La detección precoz de EIM se inició en la década de los años 60 con el desarrollo a partir de una gota de sangre recogida en papel de filtro y mediante una prueba bacteriológica capaz de identificar la elevación de los niveles de fenilalanina (test de Guthrie). El conocimiento temprano del trastorno permitió el tratamiento precoz de la fenilcetonuria (PKU) mediante restricción dietética del aminoácido fenilalanina. La ampliación del cribado metabólico neonatal más allá de la PKU se ha producido en gran parte como resultado de la introducción de la espectrometría de masas en tándem, que permite analizar múltiples metabolitos en una sola muestra de sangre.(6,7)
El objetivo del cribado neonatal de EIM es la detección, diagnóstico y tratamiento precoz y el seguimiento de los recién nacidos con estas patologías para evitar las lesiones y las discapacidades asociadas a la enfermedad.(8,9)
Las enfermedades que se mencionan a continuación, tienen en común el hecho de tratarse de trastornos hereditarios del metabolismo intermediario con una expresión a nivel renal importante derivándose en nefropatías propias:
• Enfermedad de Fabry: es una enfermedad de depósito hereditaria rara ligada al cromosoma X, causada por mutaciones del gen GLA que codifica el enzima lisosomal a-galactosidasa A (a-GAL A). Se producen depósitos renales de glucoesfingolípidos en todos los compartimentos tisulares, preferentemente en los podocitos, pero también en mesangio, endotelio del capilar glomerular, epitelio tubular, células endoteliales y de la capa muscular de arterias y arteriolas, así como en las células intersticiales. Estos depósitos pueden aparecer ya en la etapa fetal. El patrón histopatológico más característico en estadíos avanzados es el de glomeruloesclerosis focal y segmentaria.
• Enfermedad de Gaucher: déficit de actividad de la glucocerebrosidasa (beta-glucosidasa, codificada por el gen GBA) que condiciona un acumulo de glicoesfingolípidos en células del sistema retículoendotelial. Existen 3 subtipos de la enfermedad y se ha descrito afectación renal en los tipos 1 y 2. Se encuentra un depósito de células de Gaucher en el glomérulo e intersticio.
• Enfermedad de Refsum: déficit de la fitanoil-CoA hidroxilasa (codificada por el gen PHYH). Caracterizada por una tétrada de anomalías clínicas: retinitis pigmentosa, neuropatía periférica, ataxia cerebelosa y niveles elevados de proteínas en el líquido cefalorraquídeo sin un aumento en el número de células. Con poca frecuencia los pacientes desarrollan tubulopatía proximal.
• Enfermedad de Von Gierke: déficit de glucosa 6-fosfatasa o de sus proteínas transportadoras, ocasionando el depósito de glucógeno en diversos órganos. Existen 12 tipos de glucogenosis pero solo el tipo I afecta al riñón. Las manifestaciones clínicas incluyen retraso del crecimiento, hepatomegalia, hipoglucemia, acidosis láctica, hiperuricemia, osteoporosis e hiperlipidemia. El glucógeno se deposita en los túbulos proximales y distales originando vacuolas en las células tubulares y un trastorno funcional semejante al síndrome de Fanconi con aminoaciduria, glucosuria y fosfaturia. También puede presentarse acidosis tubular distal, hiperuricemia, nefrolitiasis y nefrocalcinosis. Pero la anomalía más grave es la glomeruloesclerosis segmentaria y focal, en la que un periodo de hiperfiltración inicial asintomático precede a la proteinuria y posible evolución a insuficiencia renal terminal.
• Deficiencia Familiar de Lecitin-Colesterol Aciltransferasa (LCAT): trastorno del metabolismo de las lipoproteínas y causa una tríada típica de opacidades corneales difusas, anemia hemolítica de células diana y proteinuria con insuficiencia renal.
• Nefrosialidosis: tipo de oligosacaridosis (gen causante: NEU1) en la que se desarrolla una nefropatía glomerular congénita y causa la muerte a una edad temprana. Las características clínicas y radiológicas son facies dismórficas, organomegalia por depósito, retraso mental temprano y grave, y anomalias esqueléticas. Se hallan células espumosas en la médula ósea y se identifica una mancha roja cereza en el fondo de ojo.(10)
Estos son solamente algunos de los casos que se pueden comportar con un patrón renal característico, siendo a veces desapercibidos.
Gracias al desarrollo actual de nuevas herramientas para la detección de anomalías renales, que en algunos casos permiten su evaluación desde la etapa prenatal, han aumentado sus tasas de diagnóstico. Consecuentemente, esto ha contribuido a aun mayor conocimiento sobre estas patologías, incluyendo en muchos casos, el reconocimiento de la etiología genética causante de estas anomalías.(11)
El acceso a estudios metabólicos para patologías renales de origen genético, permite no solo confirmar diagnósticos específicos, sino también poder brindar estrategias de manejo y seguimiento, conocimiento sobre el pronóstico y prevención de complicaciones. Adicionalmente, permite proveer asesoramiento la familia.(12)
Los EIM, individualmente son desórdenes raros, teniendo una incidencia < 1/100,000 en muchos de ellos, pero cuando se evalúan en forma colectiva llegan a tener una frecuencia de 1/500 - 1/1500 según la zona geográfica donde se evalúe.
La mayor parte de los datos sobre la frecuencia de su presentación, provienen de información generada por los sistemas de tamiz neonatal ampliado de los países desarrollados. Estas frecuencias tienen importantes variaciones regionales y poblacionales, que son la muestra de la enorme diversidad genética del ser humano.(13)
En 1983 se inició en Cuba la detección neonatal de EIM, específicamente de fenilcetonuria, utilizando sangre seca en papel de filtro para medir la concentración de fenilalanina, por el método de Guthrie-Susi. En 1986 se extendió a todo el país el programa de pesquisa neonatal, y en el 2000 se introdujo una nueva tecnología en el pesquisaje a partir del uso del estuche diagnóstico UMTEST-PKU de producción cubana, a través del Sistema Ultra Micro Analítico (SUMA). En 1987 se dio inicio al pesquisaje de hipotiroidismo congénito a partir de sangre del cordón umbilical en el momento del nacimiento, en el 2006 se añadió a la pesquisa neonatal de fenilcetonuria a través del SUMA, el de tres nuevas enfermedades: la deficiencia de biotinidasa, galactosemia e hiperplasia adrenal congénita; en todos los casos, por tratarse de enfermedades genéticas, el diagnóstico preciso es indispensable para un adecuado asesoramiento genético a la familia.(14,15)
Los estudios de tamizaje en recién nacidos permiten el diagnóstico en un grupo de estas entidades genéticas del metabolismo, sobre todo en las más frecuentes y tratables. Sin embargo, se ha podido comprobar que sus formas benignas en ocasiones no se detectan con esas pruebas en ese momento de la vida y constituyen posteriormente un reto para el médico, cuando debutan en el niño mayor o incluso en el adulto.(16)
Se puede hacer una aproximación diagnóstica con escasos exámenes complementarios disponibles en muchos hospitales en muchas enfermedades metabólicas que se manifiestan en el período neonatal.(17)
De ahí surge el objetivo general de esta investigación: ofrecer una alternativa de cribado útil, idónea, sencilla y confiable como herramienta para el diagnóstico de nefropatías metabólicas hereditarias.
MÉTODOS
Se realizó un estudio observacional y transversal en el periodo comprendido de septiembre 2021 a febrero 2023, en el Hospital General Docente Abel Santamaría Cuadrado, provincia Pinar del Río, Cuba.
El universo estuvo constituido por 90 pacientes que cumplieron con los criterios de inclusión:
· Recién nacidos, cuyos padres dieron el consentimiento para su inclusión en el estudio.
· Con sintomatología inespecífica, historial sintomático familiar o consanguinidad parental.
Se excluyeron aquellos pacientes cuyas historias clínicas no contaron con los datos requeridos para realizar la investigación y aquellos cuyos padres no otorgaron el consentimiento para participar en el estudio.
Teniendo en cuenta esta población, en la investigación se trabajó con muestreo aleatorio, usando la fórmula:
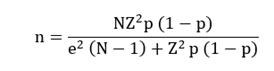
Donde:
N = es el tamaño de la población definida.
n = es el tamaño de la muestra a obtener.
Z = es el valor obtenido mediante niveles de confianza, 90 % (1,645), siendo confiable.
p = probabilidad de ocurrencia de 0,5.
e = representa el límite aceptable de error muestral, siendo 5 % (0,5) el empleado.
Al aplicar la fórmula con población conocida o finita obtenemos una cantidad de n=63 pacientes. Siendo estos, sujeto de estudio.
Las variables de estudio fueron:
Variables cuantitativas:
a) Glicemia: >45 mg/dL; <45 mg/dL (<2,2 mmol/L).
b) Lactinemia: >2,1mmol/L; <2,1mmol/L (<19mg/dl).
c) Amonemia: >150 μmol/L; <150 μmol/L (<270 μg/dl).
Variables cualitativas:
d) Hemogasometría arterial: acidosis metabólica; alcalosis metabólica; acidosis respiratoria; alcalosis respiratoria; trastorno mixto, normal.
e) Uroanálisis: sobrenadante alterado, sedimento alterado, normal.
f) Hiperazoemia: si; no
g) Prueba del talón (pesquisaje neonatal): positivo; negativo.
h) Factores de riesgo perinatales: hipoxia perinatal, prematuridad (antes de las 37 semanas de gestación), bajo peso al nacer (menos de 2500 gramos), parto distócico, enfermedades propias del embarazo, edad materna avanzada.
Los métodos utilizados fueron:
Se emplearon métodos empíricos: la observación para el diagnóstico del problema y el criterio de expertos por el método Delphi. Como métodos teóricos se utilizaron los de análisis-síntesis, inductivo–deductivo, modelación e histórico-lógico.
Del nivel estadístico se confeccionaron tablas y gráficos de distribución de frecuencia para las diferentes variables. Los cálculos se realizaron a través del programa estadístico Microsoft Excel, con el objetivo de determinar la relación entre variables y el comportamiento de muestras relacionadas con un nivel de significación (p < 0,05).
Los autores declaran que el presente estudio fue aprobado por el Consejo Científico de las instituciones participantes. La investigación se realizó conforme a los principios de la ética médica, la Declaración de Helsinki. Se procedió según las normas éticas institucionales y nacionales vigentes.
RESULTADOS
Predominó la correlación hipoglicemia, hiperlactinemia e hiperamonemia con una incidencia del 55,56 %. (Tabla 1)
|
Tabla 1. Distribución de los pacientes según la correlación entre glicemia, lactinemia y amonemia, Hospital General Docente Abel Santamaría Cuadrado, 2021-2023 |
||||
|
Amonemia |
Lactinemia |
Glicemia |
Nº |
% |
|
<150 μmol/L |
<2,1mmol/L |
<45 mg/dL |
2 |
3,17 % |
|
<150 μmol/L |
<2,1mmol/L |
>45 mg/dL |
1 |
1,59 % |
|
<150 μmol/L |
>2,1mmol/L |
<45 mg/dL |
9 |
14,29 % |
|
>150 μmol/L |
<2,1mmol/L |
<45 mg/dL |
2 |
3,17 % |
|
>150 μmol/L |
<2,1mmol/L |
>45 mg/dL |
9 |
14,29 % |
|
>150 μmol/L |
>2,1mmol/L |
<45 mg/dL |
35 |
55,56 % |
|
>150 μmol/L |
>2,1mmol/L |
>45 mg/dL |
5 |
7,94 % |
|
Total |
63 |
100 % |
||
Se encontró predominio de pacientes con acidosis metabólica en un 49,21 %, seguido en órden de frecuencia por trastornos mixtos, para un 25,40 %, respectivamente (figura 1).
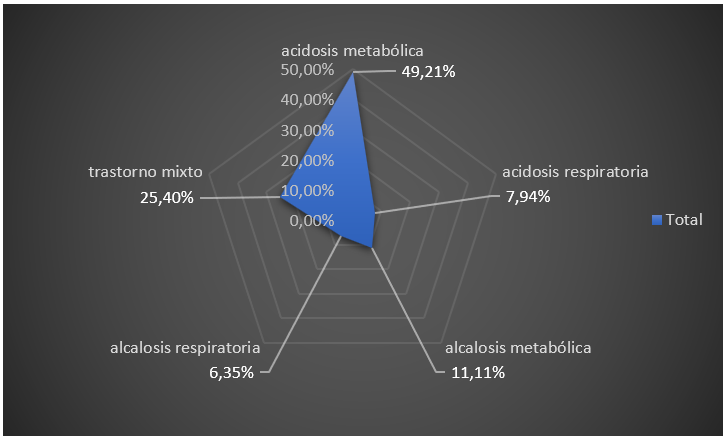
Figura 1. Distribución de los pacientes según estado hemo-gasométrico
Se observó una mayor frecuencia en la correlación de pacientes con alteraciones en el sobrenadante urinario e hiperazoemia, para un 33,33 % de la muestra (tabla 2).
|
Tabla 2. Distribución de los pacientes según correlación entre uroanálisis e hiperazoemia |
|||
|
Uroanálisis |
Hiperazoemia |
Nº |
% |
|
Normal |
No |
1 |
1,59 % |
|
Normal |
Si |
10 |
15,87 % |
|
Sedimento Alterado |
No |
10 |
15,87 % |
|
Sedimento Alterado |
Si |
12 |
19,05 % |
|
Sobrenadante Alterado |
No |
9 |
14,29 % |
|
Sobrenadante Alterado |
Si |
21 |
33,33 % |
|
Total |
63 |
100 % |
|
Fue superior el número de pacientes con pesquisa neonatal negativa, para un 87 % del total (figura 2).
En nuestro estudio, el bajo peso al nacer y la prematuridad fueron los factores de riesgo perinatal que más se asociaron a hiperazoemia en los pacientes atendidos, para un 36,51 % y 33,33 %, respectivamente, seguidos en orden de frecuencia por la hipoxia perinatal (11,11 %) (figura 3).
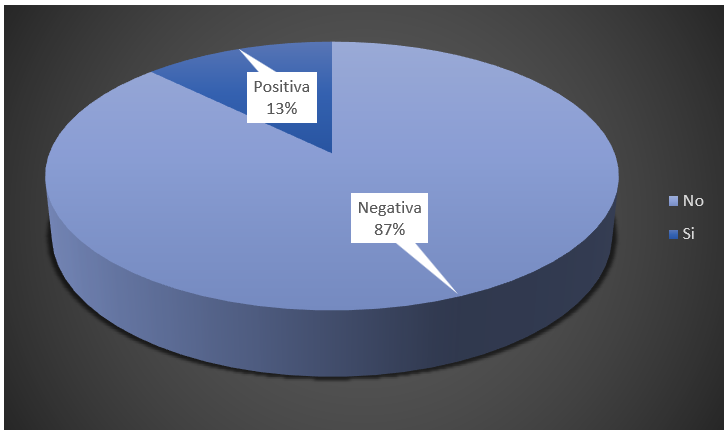
Figura 2. Distribución de los pacientes según resultado de la prueba del talón (pesquisa neonatal)
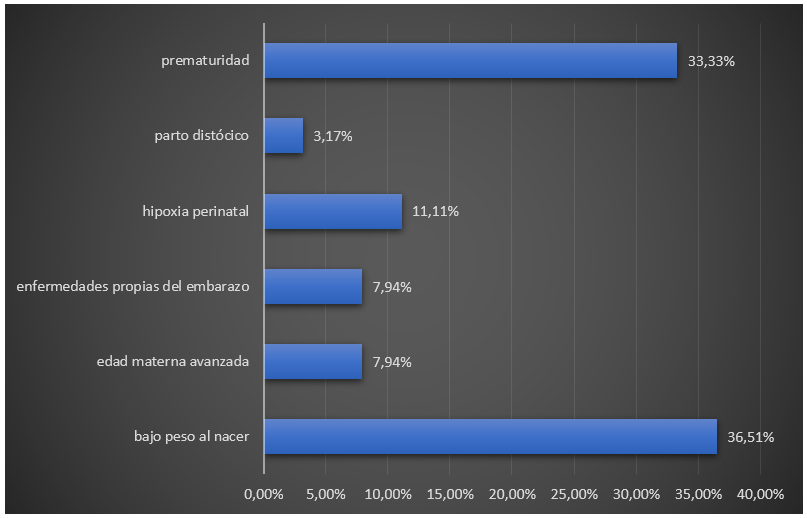
Figura 3. Distribución de los pacientes según presencia de factores de riesgo perinatales relacionados con hiperazoemia
DISCUSIÓN
Este estudio tomó como punto de partida a neonatos con sintomatología inespecífica, al igual que refleja Vargas Díaz J, considerando que el recién nacido tiene un repertorio limitado de respuestas frente a enfermedades graves y los síntomas inespecíficos iniciales incluyen rechazo de la alimentación, vómitos explosivos, progresivo compromiso de conciencia, que comienza con letargia y somnolencia y termina en coma profundo, convulsiones, compromiso hemodinámico y muerte.
En algunas otras, con síntomas crónicos y progresivos de fallo de medro, debilidad y retraso del neurodesarrollo. La disfunción de órganos por acúmulo de sustratos en ellos puede ser una variante de presentación o signos acompañantes, cardiomegalia, hepatomegalia, alteraciones esqueléticas e incluso subluxación del cristalino.(16)
Los antecedentes de consanguinidad parental, también fueron considerados, al tratarse de trastornos en su mayoría con herencia autosómica recesiva coincidiendo con las investigaciones realizadas por Couce Pico ML, et al, en las que además se tiene en cuenta la historia familiar de enfermedades metabólicas en otros miembros y muertes inexplicadas, sobre todo en hermanos.(18,19)
Según Vitoria Miñana I, et al, habitualmente en las familias no se encuentran otros casos de enfermedad (debido al menor número de hijos actual por familia, a mutaciones de novo, etc.). También se deben valorar los antecedentes familiares de encefalopatías no filiadas, y cualquier otro síndrome clínico no definido debe hacernos sospechar. No obstante, la ausencia de antecedentes familiares de interés no tiene por qué hacernos pensar que no se trata de un EIM.(20)
Los resultados del estudio concuerdan con la bibliografía analizada en cuanto a predominio de hipoglicemia, hiperlactinemia e hiperamonemia en la mayoría de los casos que se asocian a disfunción renal.
Aldámiz-Echevarría Azuara L, et al., plantea que la hipoglucemia es la alteración metabolica más común en los recién nacidos y su principal causa es el hiperinsulinismo. La hipoglucemia neonatal persistente se relaciona sobre todo con cuadros de hiperinsulinismo congénito, un grupo clínico y genéticamente heterogéneo de diferentes patologías. El exceso de insulina estimula la utilización/captación periférica de glucosa e inhibe la producción de sustratos energéticos alternativos, con la consiguiente hipoglucemia grave y persistente, pre y postprandial.
La hiperamonemia es uno de los principales síntoma bioquímicos de descompensación metabólica y se produce de forma secundaria ocasionada por la acumulación en la mitocondria, de cualquiera de los ácidos orgánicos generados por el defecto metabólico; siendo las enfermedades del ciclo de la urea y las acidemias orgánicas las causas más frecuentes de las formas graves.(21)
La elevación de lactato, indica una producción anormal de energía mitocondrial, no identifica causa. Entre las etiologías metabólicas de hiperlactinemia se pueden mencionar: algunas acidurias orgánicas, defectos gluconeogénicos, trastornos de la degradación del glucógeno, defectos de la enzima Piruvato deshidrogenasa, trastorno de cadena respiratoria y defectos en la oxidación de ácidos grasos.
El ácido láctico puede aumentar modestamente en acidurias orgánicas, a menudo la acidosis profunda y la brecha aniónica muy elevada, es atribuible a la acumulación del ácido orgánico.(22)
La acidosis metabólica severa es la alteración más frecuente del medio interno, en los recién nacidos y en la mayoría de paciente críticos. Generalmente, es debida a una acidosis láctica secundaria. Se define por: pH <7,35 y HCO3- <21 mmol/L. Es el resultado de un exceso en la producción de ácidos, por un problema en la excreción de los mismos o por una pérdida renal o gastrointestinal de bicarbonato. Los EIM relacionados con acidosis metabólica son: acidemias orgánicas, trastornos de la cadena respiratoria, trastornos del metabolismo del piruvato, Algunas enfermedades de depósito de glucógeno.
Un cálculo del Anion Gap o brecha aniónica, nos ayuda a determinar el origen de la acidosis en un paciente:
El Anion Gap normal, en una acidosis metabólica, está asociado a una pérdida gastrointestinal de bicarbonato o a trastornos de la función tubular renal que impiden o dificultan la excreción de ácidos endógenos y la reabsorción de bicarbonato. Generalmente, la acidosis es hiperclorémica.
El Anion Gap elevado, en una acidosis metabólica, indica una sobreproducción de ácidos endógenos con o sin incremento de lactato. Si no se identifica una causa evidente, se debe considerar la posibilidad de un EIM.(23)
En concordancia con los resultados de esta investigación, Ibarra-González I, et al., plantea que entre los principales datos clínicos que motivan la sospecha diagnóstica de metabolopatías, se encuentran las alteraciones en los marcadores bioquímicos de función renal, manifestándose como: orina de olor extraño, acidosis tubular renal e insuficiencia renal.(24)
Entre las características del sobrenadante urinario que nos hacen sospechar en un neonato la posibilidad de EIM, se destaca en particular, el olor anormal de la orina, según estudios publicados por Raimann B Erna. En el niño agudamente enfermo encontraremos olor a azúcar quemada en la enfermedad de la orina olor a jarabe de arce, o a pie sudado en la acidemia isovalérica y en la aciduria glutárica tipo II.(25)
Otras alteración que se manifiesta es la coloración anormal de la orina como el típico caso de la alcaptonuria, la más “aparente” enfermedad rara, ya que la orina se ennegrece inmediatamente después de su emisión debido a la oxidación del ácido homogentísico que se elimina, fruto del metabolismo de la tirosina.(26)
Las tubulopatías, típicas en estos pacientes, pueden acompañarse de nefrocalcinosis y litiasis renal y su principal forma de manifestación son las acidosis tubulares renales (ATR), dada por la presencia de ácidos orgánicos en orina que se deposita en los túbulos proximales y distales originando vacuolas en las células tubulares y un trastorno funcional semejante al síndrome de Fanconi con aminoaciduria, glucosuria y fosfaturia.
En la mayoría de las nefropatías metabólicas hereditarias, un periodo de hiperfiltración inicial asintomático precede a la proteinuria y posible evolución a insuficiencia renal. La elevación de azoados séricos (urea y creatinina) en el neonato no siempre se manifiesta en de forma aguda, sino que lo hace progresivamente, pudiendo manifestarse la insuficiencia renal terminal a partir de la 3ª-4ª década de la vida si no se hace un diagnóstico y tratamiento tempranos.(27,28)
La prueba del talón, como principal medio de cribado neonatal, se limita solamente a un determinado número de trastornos y no abarca el amplio espectro de posibilidades en el que se incluyen las nefropatías metabólicas hereditarias.
La punción capilar de talón es el método que se considera de elección para la toma de la muestra, debido a que representa una alternativa para extracción de volúmenes pequeños de sangre de manera rápida y sencilla, al representar una zona de gran vascularización y relativamente de poca inervación nerviosa. La punción debe realizarse dentro del área superficial más lateral externa o interna del talón, y con una profundidad máxima de 2,4 mm para evitar daño en hueso calcáneo, de acuerdo con el esquema de Blumenfeld (Figura 4).
Una muestra adecuada, puede ser portadora de información valiosa para salvar la vida de un niño (a), así como para prevenir complicaciones y discapacidad.
Existe variabilidad en los resultados dependiendo de la edad en días de vida del recién nacido, se ha probado la conveniencia de realizar esta prueba entre las 24 y 48 horas de vida para detección oportuna de anormalidades y manejo médico oportuno. Además, este tiempo representa una oportunidad para ampliar la cobertura de tamiz al posibilitar su práctica antes del egreso hospitalario del recién nacido.(29)
El análisis y discusión de los resultados de investigación, permitió el diseño de un algoritmo para el cribado diagnóstico de las metabolopatías hereditarias con expresión renal (Figura 5).
En el estudio se determinó la relación entre factores de riesgo perinatales y la hiperazoemia en los recién nacidos, encontrándose un predominio de prematuridad y bajo peso al nacer, aunque según Vitoria Miñana I, et al., en la mayoría de EIM intermediario no hay antecedentes gestacionales, neonatales y familiares de interés. La mayoría de los pacientes afectados de EIM tipo «intoxicación» son recién nacidos a término, adecuados para su edad gestacional, debido a que la placenta es capaz de detoxificar moléculas pequeñas resultantes del defecto enzimático. No obstante, pueden existir cuadros dismórficos cuando se trata de un EIM por acumulación de moléculas de mayor peso, como en los trastornos lisosomales y peroxisomales.(20)
También se puede dar el caso de falsos positivos en el cribado diagnóstico de metabolopatías debido a la influencia de alguno de los factores de riesgo perinatales antes señalados. Un ejemplo de esto, es el estudio realizado por Oliva López Y, et al. sobre hiperplasia adrenal congénita, con un predominio de casos con valores elevados de 17-hidroxiprogesterona (17OH/P) que posteriormente se normalizaron (por lo que constituyen falsos positivos), la mayoría de ellos nacidos por cesárea, parto pretérmino y con bajo peso al nacer, asociado a una disminución de la actividad de la 3-beta-hidroxiesteroide-deshidrogenasa, y cuando son menores de 30 semanas de edad gestacional aparece una disminución de la actividad de la 11-beta-deshidrogenasa o un retraso en la expresión de esta enzima, lo cual justifica la elevación de dicho metabolito. Se relaciona además con una degradación de 17OHP disminuida por inmadurez de la función hepática, a lo que se suma una producción aumentada de la hormona a causa del estrés al que están sometidos estos neonatos.(30)
Los programas de cribado del recién nacido continuarán expandiéndose para aquellos trastornos en los que una intervención temprana pueda modificar significativamente el curso de la enfermedad. Esta expansión será impulsada tanto por la demanda derivada del desarrollo de nuevas terapias para un mayor número de EIM, como de los avances tecnológicos y metodológicos de las pruebas.(31)
El conocimiento de las enfermedades metabólicas hereditarias disfruta de un crecimiento espectacular, en todos los aspectos. El avance experimentado por las ciencias biomédicas en general, y muy especialmente por la bioquímica y la biología molecular, han convertido a las que hasta hace poco eran consideradas como anomalías poco frecuentes o raras, y cuyo estudio casi solo tenía interés porque ayudaba a comprender los procesos normales del organismo humano, en el paradigma de la moderna medicina capaz de abordar el cuidado global y multifuncional de cada paciente, pero –lo más importante– de un modo completamente individualizado.
Este planteamiento, que resulta obligado en cualquier edad de la vida, cobra importancia excepcional en la edad pediátrica, porque además de que la mayoría de estas enfermedades debutan durante esa época, su posible repercusión a lo largo de toda la vida exige en todos los casos una prevención, un diagnóstico y un tratamiento, que garanticen el mejor desarrollo posible para el que el recién nacido ha sido programado.(32)
Las limitaciones de este estudio se basan precisamente en la confirmación de las sospechas diagnósticas de nefropatías metabólicas hereditarias mediante análisis genéticos, no siempre disponible. No obstante, permite orientar al médico en el seguimiento de estas patologías, guiado por un algoritmo diagnóstico de fácil comprensión.
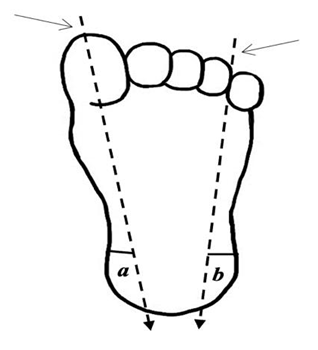
Fuente: Salmón Vega SG.(29)
Figura 4. Prueba del talón: representación esquemática de técnica de Blumenfeld
Las áreas para punción capilar de talón son bordes laterales, representados por (a) y (b). Para definirlas, se trazan dos líneas imaginarias verticales a partir de referencias indicadas con flechas, hasta lograr su proyección al talón.
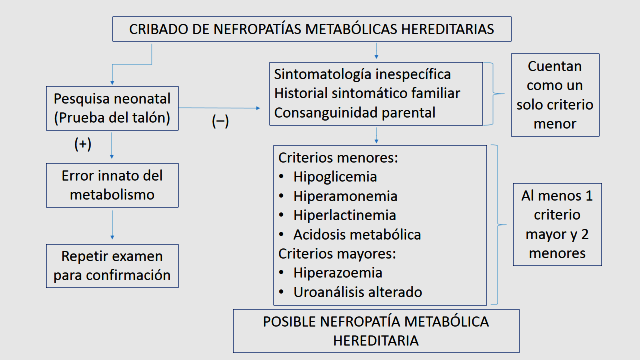
Figura 5. Algoritmo de Cribado de Nefropatías Metabólicas Hereditarias
CONCLUSIONES
En el estudio realizado, predominó la correlación hipoglicemia, hiperlactinemia e hiperamonemia con una incidencia del 55,56 % y los pacientes con acidosis metabólica en un 49,21 %. Se observó una mayor frecuencia en la correlación de pacientes con alteraciones en el sobrenadante urinario e hiperazoemia, para un 33,33 % de la muestra. Fue superior el número de pacientes con pesquisa neonatal negativa, para un 87 %. El bajo peso al nacer y la prematuridad fueron los factores de riesgo perinatales que más se asociaron a hiperazoemia en los pacientes atendidos para un 36,51 % y 33,33 % respectivamente. Los resultados obtenidos evidencian una alta sensibilidad y una baja especificidad, pero aun así nos dan un parámetro confiable y una herramienta en la ayuda del diagnóstico de nefropatías metabólicas hereditarias.
REFERENCIAS BIBLIOGRÁFICAS
1. Jeanmonod R, Asuka E, Jeanmonod D. Inborn Errors Of Metabolism. Treasure Island (FL): Stat Pearls Publishing [Internet]. 2023 Jul [citado 2024 Abr 26]. Disponible en: https://www.ncbi.nlm.nih.gov/books/NBK459183/
2. Stenton SL, Kremer LS, Kopajtich R, Ludwig C, Prokisch H. The diagnosis of inborn errors of metabolism by an integrative “multi-omics” approach: A perspective encompassing genomics, transcriptomics, and proteomics. J Inherit Metab Dis [Internet]. 2020 Ene [citado 2024 Abr 26]; 43(1):25-35. Disponible en: https://pubmed.ncbi.nlm.nih.gov/31119744/
3. López-Mejía L, Guillén-López S, Carrillo-Nieto RI, Belmont-Martínez L, Ibarra-González I, Fernández-Lainez C, Vela-Amieva M. Seguimiento bioquímico y dosis de medicamentos de EIM del metabolismo intermedio. Acta Pediatr Méx [Internet]. 2023 [citado 2024 Abr 26]; 44 (1): 75-82. Disponible en: https://doi.org/10.18233/APM44No1pp75-822573
4. Hamad ABAW, Hassan BM, Saleh AMM, Ibrahim H. Risk factors and birth prevalence of birth defects and inborn errors of metabolism in Al Ahsa, Saudi Arabia. The Pan African Medical Journal [Internet]. 23 de febrero de 2011 [citado 2024 Abr 26];8(14). Disponible en: https://www.panafrican-med-journal.com/content/article/8/14/full/
5. El-Hattab AW, Sutton VR. Inborn errors of metabolism. En: Cloherty JP, Eichenwald EC (eds). Manual of neonatal care.7th edition. Baltimore (MD): Lippincott Williams & Wilkins; 2011: 767–90.
6. Fabie NAV, Pappas KB, Feldman GL. The current state of newborn screening in the United States. Pediatr Clin North Am [Internet]. 2019 Abr [citado 2024 Abr 26]; 66(2):369-386. Disponible en: https://pubmed.ncbi.nlm.nih.gov/30819343/
7. Grupo de trabajo de protocolos de cribado neonatal de la Ponencia de cribado poblacional. Protocolo de cribado neonatal de la fenilcetonuria. Ministerio de Sanidad (Madrid); 2021.
8. Shoraka HR, Haghdoost AA, Baneshi MR, Bagherinezhad Z, Zolala F. Global prevalence of classic phenylketonuria based on Neonatal Screening Program Data: systematic review and meta-analysis. Clin Exp Pediatr [Internet]. 2020 Feb [citado 2024 Abr 26]; 63(2):34-43. Disponible en: https://pubmed.ncbi.nlm.nih.gov/32024337/
9. González-Lamuño Leguina D, Bóveda Fontán MD, Bueno Delgado M, Gort Mas L, Unceta Suárez M, Morales Conejo M. El cribado metabólico del recién nacido como modelo asistencial de la medicina de precisión. Perspectiva desde la Asociación Española para el Estudio de los Errores Congénitos del Metabolismo (AECOM). Rev Esp Salud Pública [Internet]. 2021 Ene [citado 2024 Abr 26]; 95(1):e1-e17. Disponible en: https://dialnet.unirioja.es/servlet/articulo?codigo=7957686
10. Torra Balcells R. Nefropatías por Trastorno Metabólico-Hereditario con Afectación Renal. [Internet] Nefrología al día. 2023 Feb [citado 2024 Abr 26]; Disponible en: https://www.nefrologiaaldia.org/308
11. Bekheirnia N, Glinton KE, Rossetti L, Manor J, Chen W, Lamb DJ et al. Clinical utility of genetic testing in the precision diagnosis and management of pediatric patients with kidney and urinary tract diseases. Kidney360 [Internet]. 2021 Ene [citado 2024 Abr 26]; 2(1):90-104. Disponible en: https://pubmed.ncbi.nlm.nih.gov/35368817/
12. J Lores, H Pachajoa, V Ochoa, JM Restrepo. Importancia de la genética en la nefrología pediátrica. Andes pediatr [Internet]. 2023 [citado 2024 Abr 26]; 94(4):542-553. Disponible en: https://doi:10.32641/andespediatr.v94i4.4498
13. Sanjurjo P, Baldellou A, Aldámiz-Echevarría K, Montejo M, García Jiménez MC. Los errores congénitos del metabolismo como enfermedades raras con un planteamiento global específico. Anales Sis San Navarra [Internet]. 2008 [citado 2024 Abr 27] ; 31 (2): 55-73. Disponible en: http://scielo.isciii.es/scielo.php?script=sci_arttext&pid=S1137-66272008000400005&lng=es
14. Perdomo JC, Luna E, Domínguez ME, Castro M, Rodríguez D, Landa M, Ravelo O, Monzón M. El programa de diagnóstico, manejo y prevención de enfermedades genéticas y defectos congénitos en la provincia de matanzas: 1988-2008. Rev Cubana Genet Comunit [internet]. 2009 [citado 2024 Abr 26]; 3(2,3). Disponible en: http://www.bvs.sld.cu/revistas/rcgc/v3n2_3/rcgc0523010%20esp.html
15. Marcheco B. El Programa Nacional de Diagnóstico, Manejo y Prevención de Enfermedades Genéticas y Defectos Congénitos de Cuba: 1981-2009. Rev Cubana Genet Comunit [internet]. 2009 May [citado 2024 Abr 26]; 3 (2-3): 167-84. Disponible en: http://bvs.sld.cu/revistas/rcgc/v3n2_3/cuba.pdf
16. Vargas Díaz J. Errores innatos del metabolismo. Revista Cubana de Pediatría [internet]. 2019 [citado 2024 Abr 26]; 91(3):e872. Disponible en: http://bvs.sld.cu/revistas/ped/
17. Rebage V, López-Pisón J, Baldellou A. Errores congénitos del metabolismo en el período neonatal. En: Sanjurjo P, Baldellou A (eds). Diagnóstico y tratamiento de las enfermedades metabólicas hereditarias. Madrid: Ergon; 2006: 107-24.
18. Couce Pico ML, Fernández Lorenzo JR, Fraga Bermúdez JM. Enfermedades congénitas del metabolismo en el período neonatal. En: Protocolos Diagnóstico-Terapéuticos de Neonatología de la AEP. Madrid: Ergón; 2011. p.361-369.
19. Couce Pico ML, Fraga Bermudez JM. Errores Congénitos del Metabolismo. En: Moro M, Vento M (editores). De Guardia en Neonatología 3ª Ed. Madrid: Ergón; 2016. p.198-203.
20. Vitoria Miñana I, Rausell Félix D, Lahuerta Cervera S, Sánchez Zahonero S, Dalmau Serra J. Errores innatos del metabolismo intermediario. Propuesta de guía diagnóstica de urgencias en un hospital comarcal. Acta Pediatr Esp [Internet]. 2013 [citado 2024 Jun 03]; 71(2): 47-53. Disponible en: https://actapediatrica.com/index.php/secciones/nutricion-infantil/
21. Aldámiz-Echevarría Azuara L, Couce Pico ML, González-Lamuño Leguina D, García Jiménez MC (editores). Enfermedades Raras Metabólicas. Procedimientos de Urgencias y de Situaciones de Riesgo. Madrid: Ergón; 2017.
22. Fadlallah F. Protocolo De Abordaje Diagnóstico De Errores Innatos Del Metabolismo. Hospital Del Niño Doctor José Renán Esquivel. Departamento De Neonatología. Sep; 2021.
23. Guía de Práctica Clínica para el Diagnóstico y Tratamiento de los Errores Innatos del Metabolismo. Unidad De Soporte Al Diagnóstico Y Tratamiento. Sub Unidad De Soporte Al Diagnóstico – Genética. Instituto Nacional de Salud del Niño San Borja. Oct; 2020
24. Ibarra González I, Fernández Lainez C, Belmont Martínez L, Guillén López S, Monroy Santoyo S, Vela Amieva M. Caracterización de errores innatos del metabolismo intermediario en pacientes mexicanos. An Pediatr (Barc) [Internet]. 2014 [citado 2024 Jun 15]; 80 (5): 310-316. Disponible en: https://analesdepediatria.org/es-caracterizacion-errores-innatos-del-metabolismo-articulo-S1695403313003809
25. Raimann B Erna. Diagnóstico de errores innatos del metabolismo. Rev Chil Pediatr [Internet]. 2008 Nov [citado 2024 Jun 01]; 79(1): 92-95. Disponible en: http://dx.doi.org/10.4067/S0370-41062008000700014
26. Pérez González B (coord.), Mayor Zaragoza F, Medina JM, Couce ML, Ribes A, Palau F, et al. 50 años de cribado neonatal: cómo afrontamos el futuro. Madrid: Editorial Centro de Estudios Fundación Ramón Areces; 2021.
27. Torra Balcells R. Nefropatías por Trastorno Metabólico-Hereditario con Afectación Renal. [Internet] Nefrología al día. 2023 Feb [citado 2024 Abr 26]; Disponible en: https://www.nefrologiaaldia.org/308
28. Bustos Lozano G, Orbea Gallardo C, Fernández de Miguel S, Muñoz Labián MC, López Maestro M, Moral Pumarega M. Factores determinantes de la elevación de la uremia en los primeros días de vida en recién nacidos con menos de 30 semanas de gestación. An Pediatr (Barc) [Internet]. 2003 [citado 2024 Jun 15]; 59(6):559-64. Disponible en: https://www.analesdepediatria.org/es-pdf-S1695403303787803
29. Salmón Vega SG. Intervención de enfermería en tamiz metabólico neonatal: Revisión integrativa. SANUS [Internet]. 2022 Dic [citado 2024 Jun 15]; 7: e309. Disponible en: http://www.scielo.org.mx/scielo.php?script=sci_arttext&pid=S2448-60942022000100110&lng=es
30. Oliva López Y, González García R. Programa de detección de errores innatos del metabolismo, Minas de Matahambre 2008-2012. Revista de Ciencias Médicas de Pinar del Río [Internet]. 2014 Ene-Feb [citado 2024 Jun 03]; 18(1):66-75. Disponible en: http://scielo.sld.cu/scielo.php?script=sci_arttext&pid=S1561-31942014000100008&lng=es
31. González-Lamuño Leguina D, Bóveda Fontán MD, Bueno Delgado M, Gort Mas L, Unceta Suárez M, Morales Conejo M. El cribado metabólico del recién nacido como modelo asistencial de la medicina de precisión. Perspectiva desde la Asociación Española para el Estudio de los Errores Congénitos del Metabolismo (AECOM). Rev Esp Salud Pública [Internet]. 2021 Ene [citado 2024 Jun 15]; 95 (26):e1-17. Disponible en: https://www.mscbs.gob.es/biblioPublic/publicaciones/recursos_propios/resp/revista_cdrom/vol95/originales/rs95c_202101021.pdf
32. Baldellou Vázquez A, Dalmau Sierra J, Sanjurjo Crespo P. En: LA Echevarría Azuara, ML Couce Pico, DG Lamuño Leguina (eds). Errores innatos del metabolismo. Casos clínicos en la práctica asistencial. Majadahonda (Madrid); 2023.
FINANCIACIÓN
Los autores no recibieron financiación para el desarrollo de la presente investigación.
CONFLICTO DE INTERESES
Los autores declaran que no existe conflicto de intereses.
CONTRIBUCIÓN DE LOS AUTORES
Conceptualización: Yangel Fuentes Milián, Danyer Daniel Tamayo Ribeaux, Anabel Cepero Rodríguez, Bárbara Martínez Pérez.
Curación de datos: Yangel Fuentes Milián, Danyer Daniel Tamayo Ribeaux, Anabel Cepero Rodríguez, Bárbara Martínez Pérez.
Análisis formal: Yangel Fuentes Milián, Danyer Daniel Tamayo Ribeaux, Anabel Cepero Rodríguez, Bárbara Martínez Pérez.
Investigación: Yangel Fuentes Milián, Danyer Daniel Tamayo Ribeaux, Anabel Cepero Rodríguez, Bárbara Martínez Pérez.
Metodología: Yangel Fuentes Milián, Danyer Daniel Tamayo Ribeaux, Anabel Cepero Rodríguez, Bárbara Martínez Pérez.
Administración del proyecto: Yangel Fuentes Milián, Danyer Daniel Tamayo Ribeaux, Anabel Cepero Rodríguez, Bárbara Martínez Pérez.
Redacción – borrador original: Yangel Fuentes Milián, Danyer Daniel Tamayo Ribeaux, Anabel Cepero Rodríguez, Bárbara Martínez Pérez.
Redacción – revisión y edición: Yangel Fuentes Milián, Danyer Daniel Tamayo Ribeaux, Anabel Cepero Rodríguez, Bárbara Martínez Pérez.