doi: 10.62486/agmu202467
ORIGINAL
Screening and diagnostic algorithm of hereditary metabolic nephropathies in newborns
Cribado y algoritmo diagnóstico de nefropatías metabólicas hereditarias en recién nacidos
Yangel
Fuentes Milián1 ![]() *, Danyer Daniel Tamayo
Ribeaux2
*, Danyer Daniel Tamayo
Ribeaux2 ![]() , Anabel Cepero Rodríguez3
, Anabel Cepero Rodríguez3 ![]() , Bárbara Martínez Pérez4
, Bárbara Martínez Pérez4 ![]()
1Universidad de Ciencias Médicas de Pinar del Río. Facultad de Ciencias Médicas “Dr. Ernesto Che Guevara de la Serna”. Hospital General Docente “Abel Santamaría Cuadrado”. Servicio de Nefrología. Pinar del Río, Cuba.
2Universidad de Ciencias Médicas de Granma. Facultad de Ciencias Médicas “Celia Sánchez Manduley”. Hospital Clínico-Quirúrgico Docente “Celia Sánchez Manduley”. Servicio de Medicina Interna. Granma, Cuba.
3Universidad de Ciencias Médicas de Ciego de Ávila. Facultad de Ciencias Médicas “José Assef Yara”. Departamento de Ciencias Básicas Biomédicas. Ciego de Ávila, Cuba.
4Universidad de Ciencias Médicas de Guantánamo. Facultad de Tecnología y Enfermería “Rafael García Moreaux”. Hospital General Docente “Dr. Agustino Neto”. Servicio de Laboratorio Clínico. Guantánamo, Cuba.
Cite as: Fuentes Milián Y, Tamayo Ribeaux DD, Cepero Rodríguez A, Martínez Pérez B. Screening and diagnostic algorithm of hereditary metabolic nephropathies in newborns. Multidisciplinar (Montevideo). 2024; 2:67. https://doi.org/10.62486/agmu202467
Submitted: 22-11-2023 Revised: 16-03-2024 Accepted: 30-08-2024 Published: 31-08-2024
Editor: Telmo
Raúl Aveiro-Róbalo ![]()
Corresponding Author: Yangel Fuentes Milián *
ABSTRACT
Introduction: inborn errors of metabolism expressed as hereditary nephropathies, entail various biochemical abnormalities that facilitate their screening and diagnosis in the newborn.
Objective: to offer a useful, ideal, simple and reliable screening alternative as a tool for the diagnosis of hereditary metabolic nephropathies in newborns.
Method: an observational and cross-sectional study was carried out during the period September 2021-February 2023, at the Abel Santamaría Cuadrado General Teaching Hospital, Pinar del Río province, Cuba. The universe consisted of 90 patients and a representative sample of 63 was taken. The variables were studied: glycemia, lactinemia, ammonemia, arterial hemogasometry, urinalysis, hyperazotemia, heel test and perinatal risk factors associated with hyperazotemia. Empirical, theoretical and statistical methods were used. Medical ethics were respected.
Results: the correlation predominated hypoglycemia, hyperlactinemia and hyperammonemia with an incidence of 55,56 % and patients with metabolic acidosis in 49,21 %. A greater frequency was observed in the correlation of patients with alterations in the urinary supernatant and hyperazoemia, for 33,33 % of the sample. The number of patients with negative neonatal screening was higher, at 87 %. Low birth weight and prematurity were the perinatal risk factors most associated with hyperazoemia in the patients treated, 36,51 % and 33,33 % respectively.
Conclusions: the results obtained show high sensitivity and low specificity, but they still give us a reliable parameter and a tool to help diagnose hereditary metabolic nephropathies.
Keywords: Inborn Errors of Metabolism; Screening; Nephropathies; Newborn.
RESUMEN
Introducción: los errores innatos del metabolismo expresados como nefropatías hereditarias, conllevan diversas anormalidades bioquímicas que facilitan su cribado y diagnóstico en el recién nacido.
Objetivo: ofrecer una alternativa de cribado útil, idónea, sencilla y confiable como herramienta para el diagnóstico de nefropatías metabólicas hereditarias en recién nacidos.
Método: se realizó un estudio observacional y transversal durante el periodo septiembre 2021-febrero 2023, en el Hospital General Docente Abel Santamaría Cuadrado, provincia Pinar del Río, Cuba. El universo estuvo constituido por 90 pacientes y se tomó una muestra representativa de 63. Se estudiaron las variables: glicemia, lactinemia, amonemia, hemogasometría arterial, uroanálisis, hiperazoemia, prueba del talón y factores de riesgo perinatales asociados a hiperazoemia. Se emplearon métodos empíricos, teóricos y estadísticos. Se respetó la ética médica.
Resultados: predominó la correlación hipoglicemia, hiperlactinemia e hiperamonemia con una incidencia del 55,56 % y los pacientes con acidosis metabólica en un 49,21 %. Se observó una mayor frecuencia en la correlación de pacientes con alteraciones en el sobrenadante urinario e hiperazoemia, para un 33,33 % de la muestra. Fue superior el número de pacientes con pesquisa neonatal negativa, para un 87 %. El bajo peso al nacer y la prematuridad fueron los factores de riesgo perinatales que más se asociaron a hiperazoemia en los pacientes atendidos para un 36,51 % y 33,33 % respectivamente.
Conclusiones: los resultados obtenidos evidencian una alta sensibilidad y una baja especificidad, pero aun así nos dan un parámetro confiable y una herramienta en la ayuda del diagnóstico de nefropatías metabólicas hereditarias.
Palabras clave: Errores Innatos del Metabolismo; Cribado; Nefropatías; Recién Nacido.
INTRODUCTION
Inborn errors of intermediate metabolism (IEIM) are a heterogeneous group of monogenic diseases belonging to the group of rare diseases (RD), which affect metabolic pathways involved in the synthesis or catabolism of carbohydrates, amino acids, or lipids. These diseases involve biochemical abnormalities that can be quantified in different biological fluids, allowing for diagnosis.(1,2,3)
Most IEMs are autosomal recessive (AR); a tiny group are X-linked recessive. This situation makes it difficult to detect the birth of patients with this pathology, as most of the time, they are born into healthy families with no history of metabolic disease.(4)
Various mechanisms of enzyme deficiency can cause metabolic disorders:
a) Interruption of a step in a metabolic pathway, causing accumulation of the metabolites present before this enzymatic reaction. (Phenylketonuria).
b) Inability to produce confident intermediates or end products of a specific metabolic pathway, such as ketoacidosis in the fasting phase in medium-chain acyl-CoA dehydrogenase (MCAD) deficiency.
c) Alterations of more than one enzyme affecting several metabolic steps (less common), such as multiple sulfatase or cofactor deficiency.(5)
Early detection of IEM began in the 1960s with the development of a bacteriological test capable of identifying elevated phenylalanine levels (Guthrie test) from a drop of blood collected on filter paper. Early knowledge of the disorder allowed for early phenylketonuria (PKU) treatment through dietary restriction of the amino acid phenylalanine. The expansion of neonatal metabolic screening beyond PKU has primarily resulted from the introduction of tandem mass spectrometry, which allows multiple metabolites to be analyzed in a single blood sample.(6,7)
The goal of neonatal screening for IEM is the early detection, diagnosis, treatment, and follow-up of newborns with these conditions to prevent disease-related injuries and disabilities.(8,9)
The diseases listed below have in common the fact that they are hereditary disorders of intermediate metabolism with significant renal expression, leading to specific nephropathies:
· Fabry disease: this is a rare hereditary storage disease linked to the X chromosome, caused by mutations in the GLA gene that encodes the lysosomal enzyme a-galactosidase A (a-GAL A). Glycosphingolipid deposits occur in all tissue compartments, primarily in podocytes, but also in the mesangium, glomerular capillary endothelium, tubular epithelium, endothelial cells, and the muscular layer of arteries and arterioles, as well as in interstitial cells. These deposits may already be present in the fetal stage. The most characteristic histopathological pattern in advanced stages is focal and segmental glomerulosclerosis.
· Gaucher disease: deficiency of glucocerebrosidase activity (beta-glucosidase, encoded by the GBA gene) that causes an accumulation of glycosphingolipids in cells of the reticuloendothelial system. The disease has three subtypes, and renal involvement has been described in types 1 and 2. A deposit of Gaucher cells is found in the glomerulus and interstitium.
· Refsum disease: deficiency of phytanoyl-CoA hydroxylase (encoded by the PHYH gene). Characterized by a tetrad of clinical abnormalities: retinitis pigmentosa, peripheral neuropathy, cerebellar ataxia, and elevated protein levels in the cerebrospinal fluid without an increase in cell count. Rarely do patients develop proximal tubulopathy.
· Von Gierke disease: deficiency of glucose-6-phosphatase or its carrier proteins, causing glycogen to be deposited in various organs. There are 12 glycogen storage disease types, but only type I affects the kidneys. Clinical manifestations include growth retardation, hepatomegaly, hypoglycemia, lactic acidosis, hyperuricemia, osteoporosis, and hyperlipidemia. Glycogen is deposited in the proximal and distal tubules, causing vacuoles in the tubular cells and a functional disorder similar to Fanconi syndrome with aminoaciduria, glucosuria, and phosphaturia. Distal tubular acidosis, hyperuricemia, nephrolithiasis, and nephrocalcinosis may also occur. However, the most serious abnormality is focal segmental glomerulosclerosis, in which an initial asymptomatic period of hyperfiltration precedes proteinuria and possible progression to end-stage renal failure.
· Familial lecithin-cholesterol acyltransferase (LCAT) deficiency: a disorder of lipoprotein metabolism that causes a typical triad of diffuse corneal opacities, hemolytic target cell anemia, and proteinuria with renal failure.
· Nephrosialidosis is a type of oligosaccharidosis (causative gene: NEU1) in which congenital glomerular nephropathy develops and causes death at an early age. Clinical and radiological features include dysmorphic facies, organomegaly due to deposition, early and severe mental retardation, and skeletal abnormalities. Foam cells are found in the bone marrow, and a cherry-red spot is identified at the back of the eye.(10)
These are just some of the cases that can present with a characteristic renal pattern, sometimes going unnoticed.
Thanks to the current development of new tools for detecting kidney abnormalities, which in some cases allow for evaluation from the prenatal stage, diagnosis rates have increased. Consequently, this has contributed to even greater knowledge about these pathologies, including, in many cases, recognition of the genetic etiology causing these abnormalities.(11)
Access to metabolic studies for genetic kidney diseases not only confirms specific diagnoses but also provides management and follow-up strategies, knowledge about prognosis, and prevention of complications. In addition, it allows counseling to be provided to the family.
IEMs are individually rare disorders, with an incidence of < 1/100 000 in many cases, but when evaluated collectively, they have a frequency of 1/500 - 1/1500, depending on the geographical area where they are evaluated.
Most of the data on the frequency of their occurrence comes from information generated by expanded neonatal screening systems in developed countries. These frequencies have significant regional and population variations, which are evidence of the enormous genetic diversity of human beings.(13)
In 1983, neonatal screening for IEM, specifically phenylketonuria, began in Cuba using dried blood on filter paper to measure phenylalanine concentration by the Guthrie-Susi method. In 1986, the neonatal screening program was extended to the entire country, and in 2000, a new technology was introduced for screening based on the use of the Cuban-made UMTEST-PKU diagnostic kit through the Ultra Micro Analytical System (SUMA). In 1987, screening for congenital hypothyroidism began using umbilical cord blood at birth. In 2006, three new diseases were added to the SUMA neonatal screening program for phenylketonuria: biotinidase deficiency, galactosemia, and congenital adrenal hyperplasia. In all cases, because these are genetic diseases, accurate diagnosis is essential for adequate genetic counseling for the family.(14,15)
Screening tests in newborns enable the diagnosis of genetic metabolic disorders, especially the most common and treatable ones. However, benign forms are sometimes not detected by these tests at that stage of life and pose a challenge for physicians when they first appear in older children or adults.
A diagnostic approach can be made with a few additional tests available in many hospitals for metabolic diseases manifesting in the neonatal period.(17)
Hence, the overall objective of this research is to offer a practical, suitable, simple, and reliable screening alternative to diagnose hereditary metabolic kidney diseases.
METHOD
An observational, cross-sectional study was conducted at the Abel Santamaría Cuadrado General Teaching Hospital in Pinar del Río province, Cuba, between September 2021 and February 2023.
The study population consisted of 90 patients who met the following inclusion criteria:
· Newborns whose parents gave consent for their inclusion in the study.
· With nonspecific symptoms, a family history of symptoms, or parental consanguinity.
Patients whose medical records did not contain the data required to conduct the research and those whose parents did not consent to participate were excluded. Considering this population, the research used random sampling using the formula:
![]()
Where:
N = is the size of the defined population.
n = is the size of the sample to be obtained.
Z = is the value obtained using confidence levels, 90 % (1,645), which is reliable.
p = probability of occurrence of 0,5.
e = represents the acceptable sampling error limit, with 5 % (0,5) being used.
When applying the formula with a known or finite population, we obtain a number of n=63 patients. These are the subjects of the study. The study variables were:
Quantitative variables
a) Blood glucose: >45 mg/dL; <45 mg/dL (<2,2 mmol/L).
b) Lactate: >2,1 mmol/L; <2,1 mmol/L (<19 mg/dL).
c) Ammonia: >150 μmol/L; <150 μmol/L (<270 μg/dL).
Qualitative variables
d) Arterial blood gas analysis: metabolic acidosis; metabolic alkalosis; respiratory acidosis; respiratory alkalosis; mixed disorder, normal.
e) Urine analysis: altered supernatant, altered sediment, normal.
f) Hyperazotemia: yes; no
g) Heel prick test (neonatal screening): positive; negative.
h) Perinatal risk factors: perinatal hypoxia, prematurity (before 37 weeks of gestation), low birth weight (less than 2500 grams), dystocic delivery, pregnancy-related diseases, advanced maternal age.
The methods used were:
Empirical methods were used: observation for diagnosis of the problem and expert opinion using the Delphi method. Theoretical methods included analysis-synthesis, inductive-deductive, modeling, and historical-logical.
Frequent distribution tables and graphs were prepared for the different variables at the statistical level. The calculations were performed using the statistical program Microsoft Excel to determine the relationship between variables and the behavior of samples related to a level of significance (p < 0,05).
The authors declare that the Scientific Council approved this study of the participating institutions. The research was conducted using the principles of medical ethics and the Declaration of Helsinki. Current institutional and national ethical standards carried it out.
RESULTS
The correlation between hypoglycemia, hyperlactemia, and hyperammonemia was predominant, with an incidence of 55,56 % (table 1).
|
Table 1. Distribution of patients according to the correlation between blood glucose, lactate, and ammonia levels. Abel Santamaría Cuadrado General Teaching Hospital. 2021-2023 |
||||
|
Amonemia |
Lactemia |
Blood sugar |
Nº |
% |
|
<150 μmol/L |
<2,1mmol/L |
<45 mg/dL |
2 |
3,17 |
|
<150 μmol/L |
<2,1mmol/L |
>45 mg/dL |
1 |
1,59 |
|
<150 μmol/L |
>2,1mmol/L |
<45 mg/dL |
9 |
14,29 |
|
>150 μmol/L |
<2,1mmol/L |
<45 mg/dL |
2 |
3,17 |
|
>150 μmol/L |
<2,1mmol/L |
>45 mg/dL |
9 |
14,29 |
|
>150 μmol/L |
>2,1mmol/L |
<45 mg/dL |
35 |
55,56 |
|
>150 μmol/L |
>2,1mmol/L |
>45 mg/dL |
5 |
7,94 |
|
Total |
63 |
100 |
||
A predominance of patients with metabolic acidosis was found in 49,21 %, followed in order of frequency by mixed disorders, at 25,40 %, respectively (figure 1).
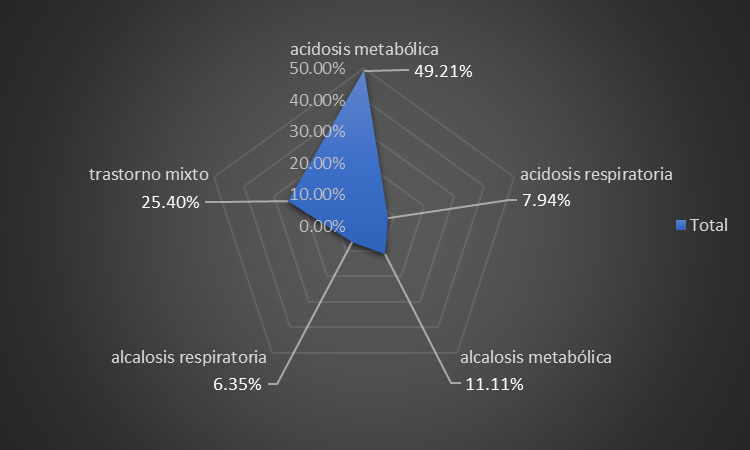
Figure 1. Distribution of patients according to blood gas status
A higher frequency was observed in the correlation of patients with alterations in urinary supernatant and hyperazoemia, accounting for 33,33 % of the sample (table 2).
|
Table 2. Distribution of patients according to correlation between urinalysis and hyperazoemia |
|||
|
Urine analysis |
Hyperazoemia |
Nº |
% |
|
normal |
No |
1 |
1,59 |
|
normal |
Yes |
10 |
15,87 |
|
altered sediment |
No |
10 |
15,87 |
|
altered sediment |
Yes |
12 |
19,05 |
|
altered supernatant |
No |
9 |
14,29 |
|
altered supernatant |
Yes |
21 |
33,33 |
|
Total |
63 |
100 |
|
The number of patients with negative neonatal screening was higher, accounting for 87 % of the total (figure 2).
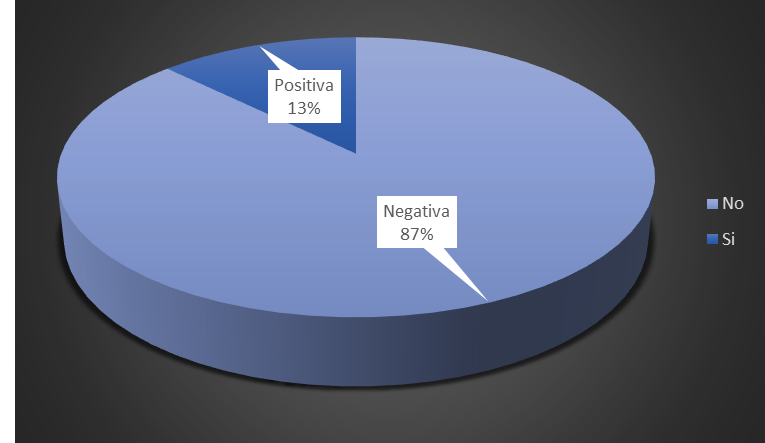
Figure 2. Distribution of patients according to heel prick test results (neonatal screening)
In our study, low birth weight and prematurity were the perinatal risk factors most associated with hyperazotemia in the patients treated, at 36,51 % and 33,33 %, respectively, followed in order of frequency by perinatal hypoxia (11,11 %) (figure 3).
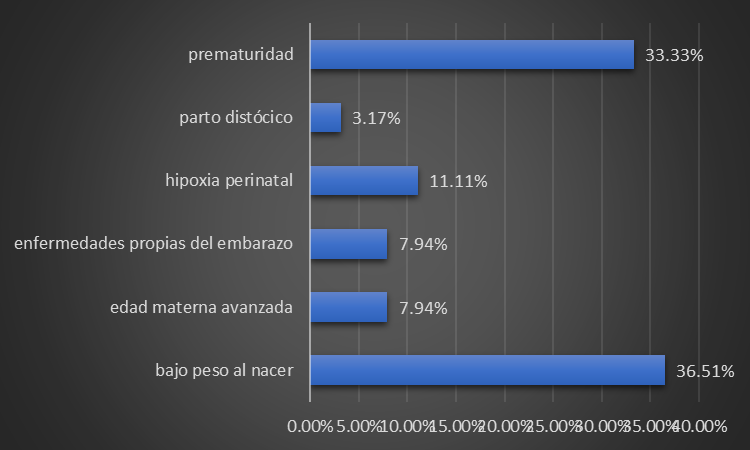
Figure 3. Distribution of patients according to the presence of perinatal risk factors related to hyperazoemia
DISCUSSION
This study took as its starting point newborns with nonspecific symptoms, as reflected by Vargas Díaz J(16), considering that newborns have a limited repertoire of responses to serious illnesses and that initial nonspecific symptoms include refusal to feed, explosive vomiting, progressive loss of consciousness, beginning with lethargy and drowsiness and ending in deep coma, seizures, hemodynamic compromise and death.
In some other cases, there are chronic and progressive symptoms of failure to thrive, weakness, and neurodevelopmental delay. Organ dysfunction due to the accumulation of substrates in the organs may be a variant of presentation or accompanying signs, such as cardiomegaly, hepatomegaly, skeletal abnormalities, and even lens subluxation.(16)
A history of parental consanguinity was also considered, as these disorders are mostly autosomal recessive, coinciding with the research carried out by Couce Pico ML et al.(18,19) which also takes into account a family history of metabolic diseases in other members and unexplained deaths, especially in siblings.
According to Vitoria Miñana I et al.(20) other cases of the disease are not usually found in families (due to the smaller number of children per family today, de novo mutations, etc.). A family history of unrelated encephalopathies should also be assessed, and any other undefined clinical syndrome should raise suspicion. However, the absence of a relevant family history does not necessarily mean that it is not an IEM.
The study’s results are consistent with the literature reviewed in terms of the predominance of hypoglycemia, hyperlactatemia, and hyperammonemia in most cases associated with renal dysfunction.
Aldámiz-Echevarría Azuara L et al.(21) suggest that hypoglycemia is the most common metabolic disorder in newborns, and its leading cause is hyperinsulinism. Persistent neonatal hypoglycemia is mainly related to congenital hyperinsulinism, a clinically and genetically heterogeneous group of different pathologies. Excess insulin stimulates peripheral glucose utilization/uptake and inhibits the production of alternative energy substrates, resulting in severe and persistent pre- and postprandial hypoglycemia.
Hyperammonemia is one of the main biochemical symptoms of metabolic decompensation and occurs secondarily due to the accumulation in the mitochondria of any of the organic acids generated by the metabolic defect; urea cycle disorders and organic acidemias are the most common causes of severe forms.
Elevated lactate indicates abnormal mitochondrial energy production but does not identify a cause. Metabolic etiologies of hyperlactatemia include certain organic acidurias, gluconeogenic defects, glycogen degradation disorders, pyruvate dehydrogenase enzyme defects, respiratory chain disorders, and fatty acid oxidation defects.
Lactic acid may increase modestly in organic acidurias, often with profound acidosis and a very high anion gap attributable to organic acid accumulation.(22)
Severe metabolic acidosis is the most common internal environment disorder in newborns and most critically ill patients. It is usually due to secondary lactic acidosis. It is defined by pH <7,35 and HCO3- <21 mmol/L. It results from excess acid production, a problem with acid excretion, or renal or gastrointestinal loss of bicarbonate. IEMs related to metabolic acidosis include organic acidemias, respiratory chain disorders, pyruvate metabolism disorders, and some glycogen storage diseases.
A calculation of the anion gap helps us determine the origin of acidosis in a patient:
The normal anion gap in metabolic acidosis is associated with gastrointestinal loss of bicarbonate or renal tubular dysfunction that prevents or hinders the excretion of endogenous acids and bicarbonate reabsorption. Acidosis is generally hyperchloric.
An elevated anion gap in metabolic acidosis indicates the overproduction of endogenous acids with or without increased lactate. If no apparent cause is identified, the possibility of an EIM should be considered.(23)
By the results of this study, Ibarra-González I et al.(3) suggest that among the primary clinical data that lead to the diagnostic suspicion of metabolic disorders are alterations in biochemical markers of renal function, manifesting as strange-smelling urine, renal tubular acidosis, and renal failure.
According to studies published by Raimann B Erna, the abnormal smell of urine stands out among the characteristics of urinary supernatants that make us suspect the possibility of IMI in a newborn. In acutely ill children, we find a burnt sugar smell in the urine in maple syrup urine disease or a maple syrup smell in isovaleric acidemia and glutaric aciduria type II.
Other alterations that manifest themselves include abnormal urine coloration, as in the typical case of alkaptonuria, the most “apparent” rare disease, since the urine turns black immediately after emission due to the oxidation of homogentisic acid, which is eliminated as a result of tyrosine metabolism.(26)
Tubulopathies, typical in these patients, may be accompanied by nephrocalcinosis and renal lithiasis and mainly manifest as renal tubular acidosis (RTA). This condition is caused by the presence of organic acids in the urine that are deposited in the proximal and distal tubules, causing vacuoles in the tubular cells and a functional disorder similar to Fanconi syndrome with aminoaciduria, glucosuria and phosphaturia.
In most hereditary metabolic nephropathies, an initial asymptomatic period of hyperfiltration precedes proteinuria and possible progression to renal failure. Elevated serum nitrogen (urea and creatinine) in newborns does not always manifest acutely but rather progressively, with end-stage renal failure possibly developing in the third or fourth decade of life if diagnosis and treatment are not early.(27,28)
The heel prick test, the main means of neonatal screening, is limited to a certain number of disorders and does not cover the wide spectrum of possibilities, including hereditary metabolic nephropathies.
The heel prick is the method of choice for sample collection because it is a quick and easy way to extract small volumes of blood from a highly vascularized and relatively uninnervated area. According to the Blumenfeld scheme, the puncture should be made in the heel’s most lateral or medial region to a maximum depth of 2,4 mm to avoid damage to the calcaneal bone (figure 4).
An adequate sample can provide valuable information to save a child’s life and prevent complications and disability.
There is variability in the results depending on the age of the newborn in days of life. The advisability of performing this test between 24 and 48 hours of life for timely detection of abnormalities and timely medical management has been proven. In addition, this time represents an opportunity to expand screening coverage by enabling its practice before the newborn is discharged from the hospital.(29)
The analysis and discussion of the research results allowed the design of an algorithm to diagnose hereditary metabolic disorders with renal expression (figure 5).
The study determined the relationship between perinatal risk factors and hyperemia in newborns, finding a predominance of prematurity and low birth weight. However, according to Vitoria Miñana I et al., in most cases of intermediate IEM, there is no relevant gestational, neonatal, or family history. Most patients affected by “intoxication” type IMI are full-term newborns, appropriate for their gestational age because the placenta is capable of detoxifying small molecules resulting from the enzyme defect. However, dysmorphic features may be present in IEM caused by accumulating heavier molecules, such as in lysosomal and peroxisomal disorders.(20)
False positives may also occur in the diagnostic screening of metabolic disorders due to the influence of some of the perinatal risk factors mentioned above. An example of this is the study conducted by Oliva López Y et al. on congenital adrenal hyperplasia, with a predominance of cases with elevated levels of 17-hydroxyprogesterone (17OH/P) that subsequently normalized (therefore constituting false positives), most of them born by cesarean section, preterm birth and low birth weight, associated with a decrease in the activity of 3-beta-hydroxysteroid dehydrogenase. When they are under 30 weeks of gestational age, there is a decrease in the activity of 11-beta-dehydrogenase or a delay in the expression of this enzyme, which justifies the elevation of this metabolite. It is also related to decreased 17OHP degradation due to immature liver function, compounded by increased hormone production due to these newborns’ stress.(30)
Newborn screening programs will continue to expand for disorders where early intervention can significantly modify the course of the disease. This expansion will be driven by demand from developing new therapies for a more significant number of IEMs and by technological and methodological advances in testing.(31)
Knowledge of inherited metabolic diseases is growing dramatically in all aspects. Advances in biomedical sciences in general, and especially in biochemistry and molecular biology, have transformed what until recently were considered rare or uncommon anomalies, whose study was of interest almost solely because it helped to understand the normal processes of the human organism, into the paradigm of modern medicine capable of addressing the comprehensive and multifunctional care of each patient, but—most importantly—in a completely individualized manner.
This approach, which is essential at any age, is critical in pediatrics because, in addition to the fact that most of these diseases first appear during childhood, their potential impact throughout life requires prevention, diagnosis, and treatment in all cases to ensure the best possible development for which the newborn has been programmed.
The limitations of this study are based precisely on the confirmation of suspected diagnoses of hereditary metabolic nephropathies through genetic analysis, which is not always available. However, it does provide guidance for physicians in the follow-up of these pathologies, guided by an easy-to-understand diagnostic algorithm.
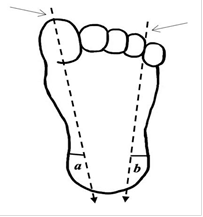
Source: Salmón Vega SG(29)
Figure 4. Heel test: schematic representation of the Blumenfeld technique
The areas for heel puncture are the lateral edges, shown in (a) and (b). To define them, draw two imaginary vertical lines from the reference points indicated by arrows until they intersect the heel.
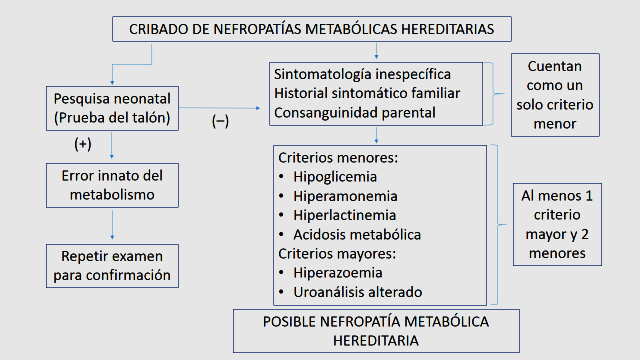
Figure 5. Algorithm for Screening Hereditary Metabolic Kidney Diseases
CONCLUSIONS
In the study conducted, the correlation between hypoglycemia, hyperlactemia, and hyperammonemia was predominant, with an incidence of 55,56 %, and patients with metabolic acidosis accounted for 49,21 %. A higher frequency was observed in the correlation of patients with alterations in urinary supernatant and hyperemia, accounting for 33,33 % of the sample. The number of patients with negative neonatal screening was higher, at 87 %. Low birth weight and prematurity were the perinatal risk factors most associated with hyperemia in the patients treated, at 36,51 % and 33,33 %, respectively. The results show high sensitivity and low specificity but provide a reliable parameter and tool to aid in diagnosing hereditary metabolic nephropathies.
BIBLIOGRAPHIC REFERENCES
1. Jeanmonod R, Asuka E, Jeanmonod D. Inborn Errors Of Metabolism. Treasure Island (FL): Stat Pearls Publishing [Internet]. 2023 Jul [citado 2024 Abr 26]. Disponible en: https://www.ncbi.nlm.nih.gov/books/NBK459183/
2. Stenton SL, Kremer LS, Kopajtich R, Ludwig C, Prokisch H. The diagnosis of inborn errors of metabolism by an integrative “multi-omics” approach: A perspective encompassing genomics, transcriptomics, and proteomics. J Inherit Metab Dis [Internet]. 2020 Ene [citado 2024 Abr 26]; 43(1):25-35. Disponible en: https://pubmed.ncbi.nlm.nih.gov/31119744/
3. López-Mejía L, Guillén-López S, Carrillo-Nieto RI, Belmont-Martínez L, Ibarra-González I, Fernández-Lainez C, Vela-Amieva M. Seguimiento bioquímico y dosis de medicamentos de EIM del metabolismo intermedio. Acta Pediatr Méx [Internet]. 2023 [citado 2024 Abr 26]; 44 (1): 75-82. Disponible en: https://doi.org/10.18233/APM44No1pp75-822573
4. Hamad ABAW, Hassan BM, Saleh AMM, Ibrahim H. Risk factors and birth prevalence of birth defects and inborn errors of metabolism in Al Ahsa, Saudi Arabia. The Pan African Medical Journal [Internet]. 23 de febrero de 2011 [citado 2024 Abr 26];8(14). Disponible en: https://www.panafrican-med-journal.com/content/article/8/14/full/
5. El-Hattab AW, Sutton VR. Inborn errors of metabolism. En: Cloherty JP, Eichenwald EC (eds). Manual of neonatal care.7th edition. Baltimore (MD): Lippincott Williams & Wilkins; 2011: 767–90.
6. Fabie NAV, Pappas KB, Feldman GL. The current state of newborn screening in the United States. Pediatr Clin North Am [Internet]. 2019 Abr [citado 2024 Abr 26]; 66(2):369-386. Disponible en: https://pubmed.ncbi.nlm.nih.gov/30819343/
7. Grupo de trabajo de protocolos de cribado neonatal de la Ponencia de cribado poblacional. Protocolo de cribado neonatal de la fenilcetonuria. Ministerio de Sanidad (Madrid); 2021.
8. Shoraka HR, Haghdoost AA, Baneshi MR, Bagherinezhad Z, Zolala F. Global prevalence of classic phenylketonuria based on Neonatal Screening Program Data: systematic review and meta-analysis. Clin Exp Pediatr [Internet]. 2020 Feb [citado 2024 Abr 26]; 63(2):34-43. Disponible en: https://pubmed.ncbi.nlm.nih.gov/32024337/
9. González-Lamuño Leguina D, Bóveda Fontán MD, Bueno Delgado M, Gort Mas L, Unceta Suárez M, Morales Conejo M. El cribado metabólico del recién nacido como modelo asistencial de la medicina de precisión. Perspectiva desde la Asociación Española para el Estudio de los Errores Congénitos del Metabolismo (AECOM). Rev Esp Salud Pública [Internet]. 2021 Ene [citado 2024 Abr 26]; 95(1):e1-e17. Disponible en: https://dialnet.unirioja.es/servlet/articulo?codigo=7957686
10. Torra Balcells R. Nefropatías por Trastorno Metabólico-Hereditario con Afectación Renal. [Internet] Nefrología al día. 2023 Feb [citado 2024 Abr 26]; Disponible en: https://www.nefrologiaaldia.org/308
11. Bekheirnia N, Glinton KE, Rossetti L, Manor J, Chen W, Lamb DJ et al. Clinical utility of genetic testing in the precision diagnosis and management of pediatric patients with kidney and urinary tract diseases. Kidney360 [Internet]. 2021 Ene [citado 2024 Abr 26]; 2(1):90-104. Disponible en: https://pubmed.ncbi.nlm.nih.gov/35368817/
12. J Lores, H Pachajoa, V Ochoa, JM Restrepo. Importancia de la genética en la nefrología pediátrica. Andes pediatr [Internet]. 2023 [citado 2024 Abr 26]; 94(4):542-553. Disponible en: https://doi:10.32641/andespediatr.v94i4.4498
13. Sanjurjo P, Baldellou A, Aldámiz-Echevarría K, Montejo M, García Jiménez MC. Los errores congénitos del metabolismo como enfermedades raras con un planteamiento global específico. Anales Sis San Navarra [Internet]. 2008 [citado 2024 Abr 27] ; 31 (2): 55-73. Disponible en: http://scielo.isciii.es/scielo.php?script=sci_arttext&pid=S1137-66272008000400005&lng=es
14. Perdomo JC, Luna E, Domínguez ME, Castro M, Rodríguez D, Landa M, Ravelo O, Monzón M. El programa de diagnóstico, manejo y prevención de enfermedades genéticas y defectos congénitos en la provincia de matanzas: 1988-2008. Rev Cubana Genet Comunit [internet]. 2009 [citado 2024 Abr 26]; 3(2,3). Disponible en: http://www.bvs.sld.cu/revistas/rcgc/v3n2_3/rcgc0523010%20esp.html
15. Marcheco B. El Programa Nacional de Diagnóstico, Manejo y Prevención de Enfermedades Genéticas y Defectos Congénitos de Cuba: 1981-2009. Rev Cubana Genet Comunit [internet]. 2009 May [citado 2024 Abr 26]; 3 (2-3): 167-84. Disponible en: http://bvs.sld.cu/revistas/rcgc/v3n2_3/cuba.pdf
16. Vargas Díaz J. Errores innatos del metabolismo. Revista Cubana de Pediatría [internet]. 2019 [citado 2024 Abr 26]; 91(3):e872. Disponible en: http://bvs.sld.cu/revistas/ped/
17. Rebage V, López-Pisón J, Baldellou A. Errores congénitos del metabolismo en el período neonatal. En: Sanjurjo P, Baldellou A (eds). Diagnóstico y tratamiento de las enfermedades metabólicas hereditarias. Madrid: Ergon; 2006: 107-24.
18. Couce Pico ML, Fernández Lorenzo JR, Fraga Bermúdez JM. Enfermedades congénitas del metabolismo en el período neonatal. En: Protocolos Diagnóstico-Terapéuticos de Neonatología de la AEP. Madrid: Ergón; 2011. p.361-369.
19. Couce Pico ML, Fraga Bermudez JM. Errores Congénitos del Metabolismo. En: Moro M, Vento M (editores). De Guardia en Neonatología 3ª Ed. Madrid: Ergón; 2016. p.198-203.
20. Vitoria Miñana I, Rausell Félix D, Lahuerta Cervera S, Sánchez Zahonero S, Dalmau Serra J. Errores innatos del metabolismo intermediario. Propuesta de guía diagnóstica de urgencias en un hospital comarcal. Acta Pediatr Esp [Internet]. 2013 [citado 2024 Jun 03]; 71(2): 47-53. Disponible en: https://actapediatrica.com/index.php/secciones/nutricion-infantil/
21. Aldámiz-Echevarría Azuara L, Couce Pico ML, González-Lamuño Leguina D, García Jiménez MC (editores). Enfermedades Raras Metabólicas. Procedimientos de Urgencias y de Situaciones de Riesgo. Madrid: Ergón; 2017.
22. Fadlallah F. Protocolo De Abordaje Diagnóstico De Errores Innatos Del Metabolismo. Hospital Del Niño Doctor José Renán Esquivel. Departamento De Neonatología. Sep; 2021.
23. Guía de Práctica Clínica para el Diagnóstico y Tratamiento de los Errores Innatos del Metabolismo. Unidad De Soporte Al Diagnóstico Y Tratamiento. Sub Unidad De Soporte Al Diagnóstico – Genética. Instituto Nacional de Salud del Niño San Borja. Oct; 2020
24. Ibarra González I, Fernández Lainez C, Belmont Martínez L, Guillén López S, Monroy Santoyo S, Vela Amieva M. Caracterización de errores innatos del metabolismo intermediario en pacientes mexicanos. An Pediatr (Barc) [Internet]. 2014 [citado 2024 Jun 15]; 80 (5): 310-316. Disponible en: https://analesdepediatria.org/es-caracterizacion-errores-innatos-del-metabolismo-articulo-S1695403313003809
25. Raimann B Erna. Diagnóstico de errores innatos del metabolismo. Rev Chil Pediatr [Internet]. 2008 Nov [citado 2024 Jun 01]; 79(1): 92-95. Disponible en: http://dx.doi.org/10.4067/S0370-41062008000700014
26. Pérez González B (coord.), Mayor Zaragoza F, Medina JM, Couce ML, Ribes A, Palau F, et al. 50 años de cribado neonatal: cómo afrontamos el futuro. Madrid: Editorial Centro de Estudios Fundación Ramón Areces; 2021.
27. Torra Balcells R. Nefropatías por Trastorno Metabólico-Hereditario con Afectación Renal. [Internet] Nefrología al día. 2023 Feb [citado 2024 Abr 26]; Disponible en: https://www.nefrologiaaldia.org/308
28. Bustos Lozano G, Orbea Gallardo C, Fernández de Miguel S, Muñoz Labián MC, López Maestro M, Moral Pumarega M. Factores determinantes de la elevación de la uremia en los primeros días de vida en recién nacidos con menos de 30 semanas de gestación. An Pediatr (Barc) [Internet]. 2003 [citado 2024 Jun 15]; 59(6):559-64. Disponible en: https://www.analesdepediatria.org/es-pdf-S1695403303787803
29. Salmón Vega SG. Intervención de enfermería en tamiz metabólico neonatal: Revisión integrativa. SANUS [Internet]. 2022 Dic [citado 2024 Jun 15]; 7: e309. Disponible en: http://www.scielo.org.mx/scielo.php?script=sci_arttext&pid=S2448-60942022000100110&lng=es
30. Oliva López Y, González García R. Programa de detección de errores innatos del metabolismo, Minas de Matahambre 2008-2012. Revista de Ciencias Médicas de Pinar del Río [Internet]. 2014 Ene-Feb [citado 2024 Jun 03]; 18(1):66-75. Disponible en: http://scielo.sld.cu/scielo.php?script=sci_arttext&pid=S1561-31942014000100008&lng=es
31. González-Lamuño Leguina D, Bóveda Fontán MD, Bueno Delgado M, Gort Mas L, Unceta Suárez M, Morales Conejo M. El cribado metabólico del recién nacido como modelo asistencial de la medicina de precisión. Perspectiva desde la Asociación Española para el Estudio de los Errores Congénitos del Metabolismo (AECOM). Rev Esp Salud Pública [Internet]. 2021 Ene [citado 2024 Jun 15]; 95 (26):e1-17. Disponible en: https://www.mscbs.gob.es/biblioPublic/publicaciones/recursos_propios/resp/revista_cdrom/vol95/originales/rs95c_202101021.pdf
32. Baldellou Vázquez A, Dalmau Sierra J, Sanjurjo Crespo P. En: LA Echevarría Azuara, ML Couce Pico, DG Lamuño Leguina (eds). Errores innatos del metabolismo. Casos clínicos en la práctica asistencial. Majadahonda (Madrid); 2023.
FINANCING
The authors did not receive funding for the development of this research.
CONFLICT OF INTEREST
The authors declare that there is no conflict of interest.
AUTHORSHIP CONTRIBUTION
Conceptualization: Yangel Fuentes Milián, Danyer Daniel Tamayo Ribeaux, Anabel Cepero Rodríguez, Bárbara Martínez Pérez.
Data curation: Yangel Fuentes Milián, Danyer Daniel Tamayo Ribeaux, Anabel Cepero Rodríguez, Bárbara Martínez Pérez.
Formal analysis: Yangel Fuentes Milián, Danyer Daniel Tamayo Ribeaux, Anabel Cepero Rodríguez, Bárbara Martínez Pérez.
Research: Yangel Fuentes Milián, Danyer Daniel Tamayo Ribeaux, Anabel Cepero Rodríguez, Bárbara Martínez Pérez.
Methodology: Yangel Fuentes Milián, Danyer Daniel Tamayo Ribeaux, Anabel Cepero Rodríguez, Bárbara Martínez Pérez.
Project management: Yangel Fuentes Milián, Danyer Daniel Tamayo Ribeaux, Anabel Cepero Rodríguez, Bárbara Martínez Pérez.
Writing – original draft: Yangel Fuentes Milián, Danyer Daniel Tamayo Ribeaux, Anabel Cepero Rodríguez, Bárbara Martínez Pérez.
Writing – review and editing: Yangel Fuentes Milián, Danyer Daniel Tamayo Ribeaux, Anabel Cepero Rodríguez, Bárbara Martínez Pérez.